
 Die erste SotM muss selbstverständlich die Synthese meines persönlichen Lieblingsmoleküls sein: Cuban.
Die erste SotM muss selbstverständlich die Synthese meines persönlichen Lieblingsmoleküls sein: Cuban.
Dies hat zum einen rein ästhetische Gründe, zum anderen finde ich die Totalsynthese besonders raffiniert, da mittels intramolekularer Cycloaddition der Käfig gebildet wird und somit ausschließlich von 2-Cyclopentenon ausgegangen werden kann.
Die erste Cuban-Synthese wurde 1964 von Philip Eaton im Journal of the American Chemical Society publiziert. Hier wird nicht von Cyclopentenon ausgegangen, da Eaton bereits in einer früheren Ausgabe gezeigt hat, dass sich dieser Bicyclus aus Cyclopentenon darstellen lässt (s. A-E).
Darstellung des Dimers[1]
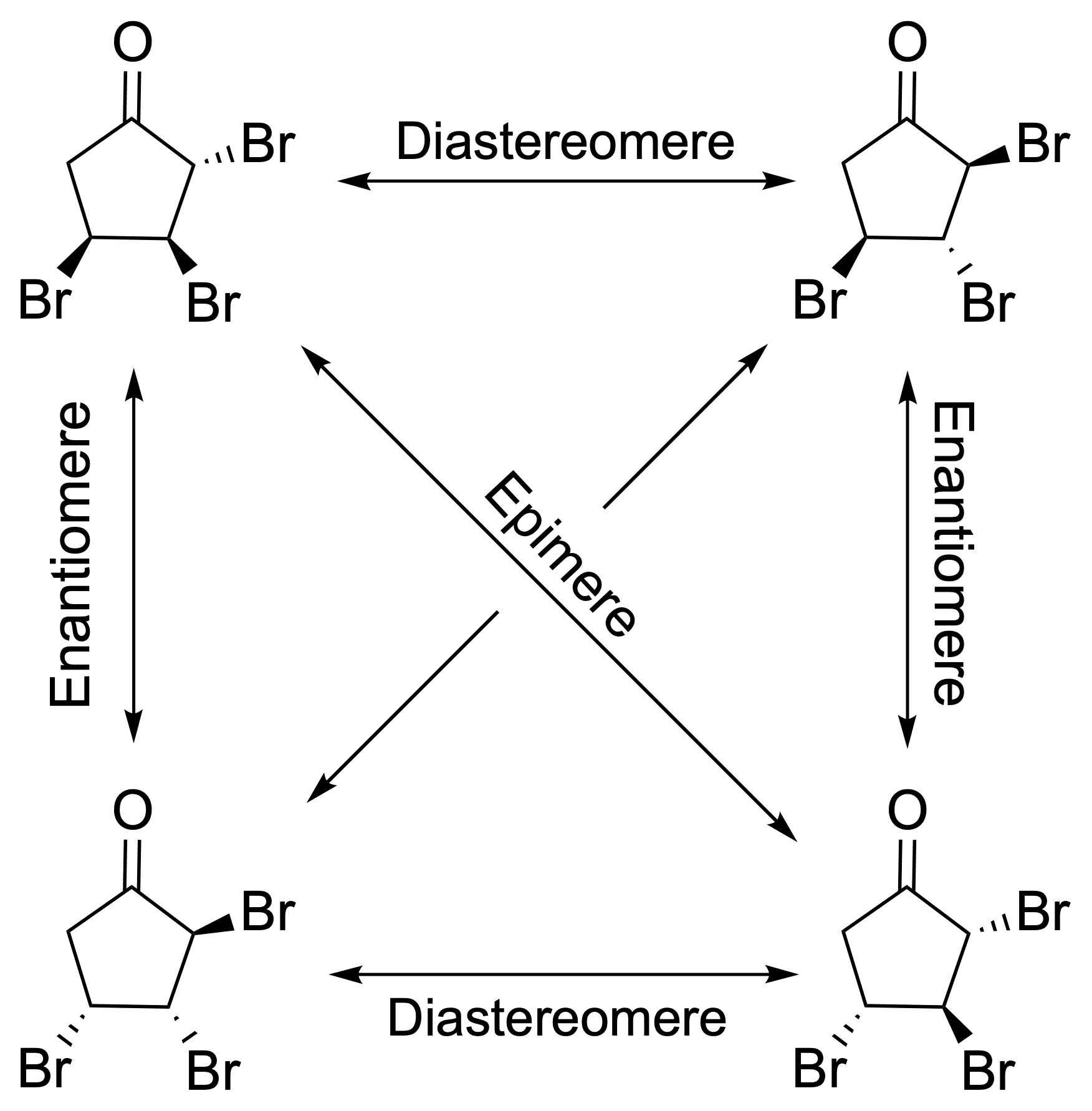
Im ersten Reaktionsschritt wird eine Wohl-Ziegler-Bromierung durchgeführt: Mit Hilfe von N-Bromsuccinimid wird in allylischer Position substituiert, ohne dass eine elektrophile Addition an der Doppelbindung stattfindet - die wird im nächsten Schritt durch die Reaktion mit elementarem Brom durchgeführt und liefert Verbindung C.Wie viele Stereoisomere können hier entstehen?
Nun wird durch Diethylamin zwei mal HBr eliminiert. Das Produkt D ist sehr kurzlebig, da es spontan via Diels-Alder-Reaktion dimerisiert. Es wird das kinetische Endo-Produkt gebildet - hier sind ebenso verschiedene Konstitutionsisomere möglich, welche im Paper kurz diskutiert werden und auf jeden Fall einer Überlegung Wert sind.

Dass hier das endo-Produkt gebildet wird, ist auf die Reaktionsbedingungen zurückzuführen. Doch warum wird Produkt 1a gegenüber Produkt 1b bevorzugt gebildet?
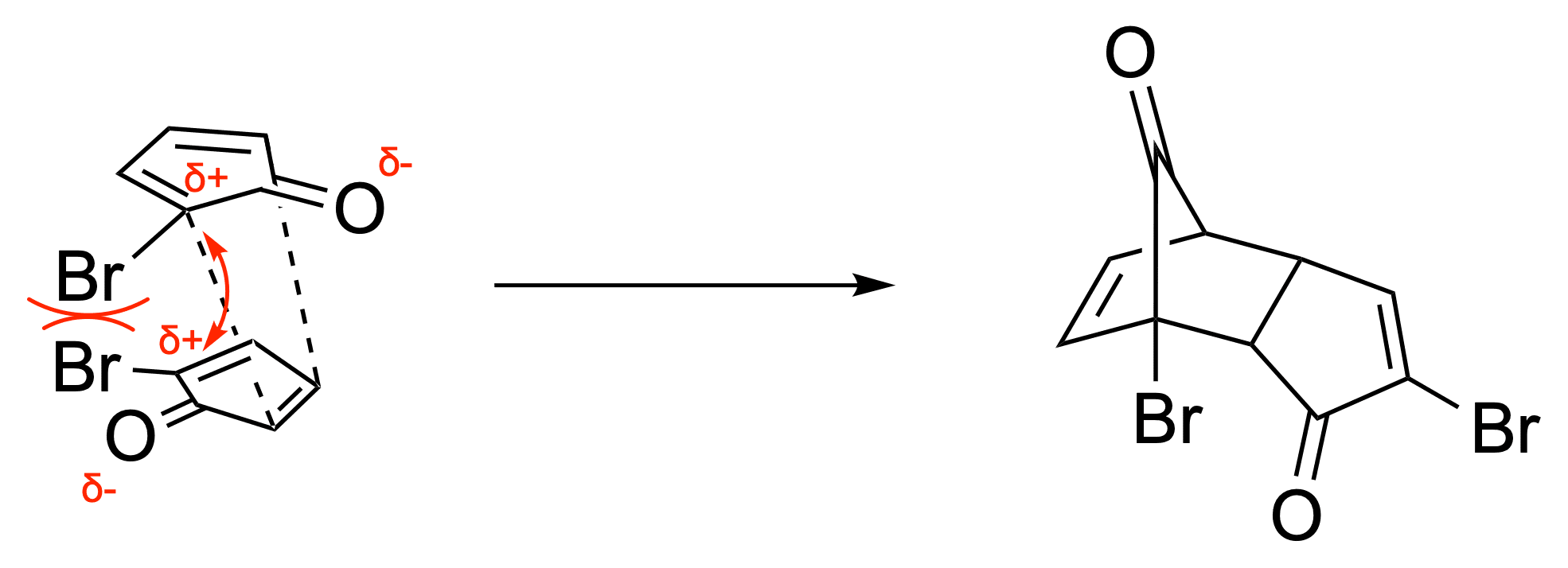
Des Weiteren dürfte auch die sterische Abstoßung durch die beiden Brom-Substituenten einen Einfluss haben.
Bildung des Käfigs[2]
Damit der Käfig geschlossen werden kann, muss eine [2+2]-Cycloaddition der beiden C-C-Doppelbindungen durchgeführt werden. Die thermische [2+2]-Cycloaddition ist symmetrieverboten, weshalb sie durch Lichtenergie induziert werden muss.Problem hierbei ist, dass dies bei Zwischenprodukt 1 zur Polymerisierung führt. Dies ist auf die Carbonyl-Gruppe der 1-C-Brücke zurückzuführen. Mittels Acetalbildung mit Ethylenglycol und para-Toluolsulfonsäure werden beide Carbonylgruppen geschützt; die konjugierte Carbonylgruppe wird im nächsten Schritt jedoch wieder entschützt:


Warum wird im zweiten Schritt nur die konjugierte Carbonylgruppe wieder selektiv entschützt?


Im Übergangszustand muss die C-O-Bindung gebrochen werden. Da Elektronendichte vom π-Orbital der Doppelbindung in das σ*-Orbital fließen kann, wird die sigma-Bindung geschwächt und kann somit leichter gebrochen werden im Übergangszustand - die Aktivierungsenergie wurde gesenkt und die Reaktion verläuft schneller als bei dem anderen Acetal, welches diesem Einfluss nicht ausgesetzt ist. Außerdem ist es auch das thermodynamisch stabilere Produkt.
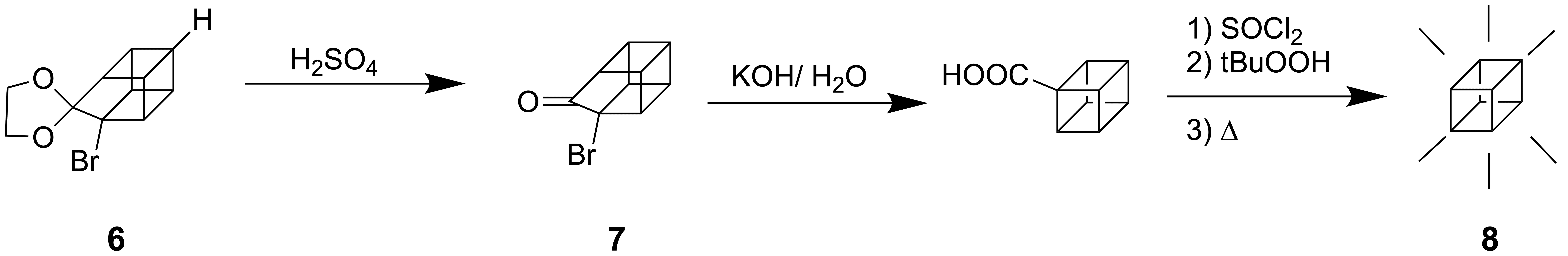
Nachdem die Cycloaddition durchgeführt wurde, steht das Grundgerüst für das Cuban-Molekül schonmal. Allerdings haben wir an zwei Kanten noch einen Kohlenstoff zu viel im Gerüst, deshalb muss eine Ringeverengung durchgeführt werden. Dies kann elegant durch eine Faworski-Umlagerung mit heißer Kalilauge durchgeführt werden. Hier wird auch ersichtlich, warum zu Beginn ein Brom-Substituent eingeführt wurde:

Jetzt sind wir unserem Würfel schon ein gutes Stück näher, es muss lediglich noch die Carbonsäuregruppe entfernt werden. Dies wird realisiert, indem die Carbonsäure mit Thionylchlorid (SOCl2) in das entsprechende Carbonsäurechlorid umgewandelt wird. Der Carbonyl-Kohlenstoff ist nun stark elektrophil und reagiert mit tert-Butylhydroperoxid zu einem Persäureester, welches unter Erhitzen in Cumol spontan radikalisch zerfällt:


Wie zu vermuten spielt die Rekombination des Alkylradikal-Intermediats mit dem tert-Butanolradikal eine große Rolle. 40% von Stoff 5 reagieren auf diesem Wege zu einem tert-Butylether. 55% reagieren auf dem obig gezeigten Weg.
Nun muss nur noch das Acetal entschützt und die Schritte von 3-7 wiederholt werden, und man erhält Cuban - wunderbar, nicht?
Referenzen
1. The Cubane System. P. Eaton, T. Cole, J. Am. Chem. Soc. 1964 86 (5), 962-964.
2. Cubane. P. Eaton, T. Cole, J. Am. Chem. Soc. 1964 86 (15), 3157-3158.
Wenn nicht anders angegeben, sind alle Abbildungen selbst angefertigt.