Klicke hier, wenn Du diesen Artikel als Video anschauen willst.×
Diesen Artikel gibt es hier auch als Video.×
 Die Wittig-Reaktion stellt einen der wichtigsten Wege dar, um Carbonyle in Alkene zu überführen. Man spricht auch von einer Olefinierung von Carbonylen. Hierbei handelt es sich im einfachsten Fall um eine Methylenierung, wie oben zu sehen; alternativ kann man durch die neue C-C-Doppelbindung auch weitere Reste einführen.
Für die Reaktion wird ein Ylid benötigt, welches auch die Stereochemie des Produkts bestimmt.
Die Wittig-Reaktion stellt einen der wichtigsten Wege dar, um Carbonyle in Alkene zu überführen. Man spricht auch von einer Olefinierung von Carbonylen. Hierbei handelt es sich im einfachsten Fall um eine Methylenierung, wie oben zu sehen; alternativ kann man durch die neue C-C-Doppelbindung auch weitere Reste einführen.
Für die Reaktion wird ein Ylid benötigt, welches auch die Stereochemie des Produkts bestimmt.
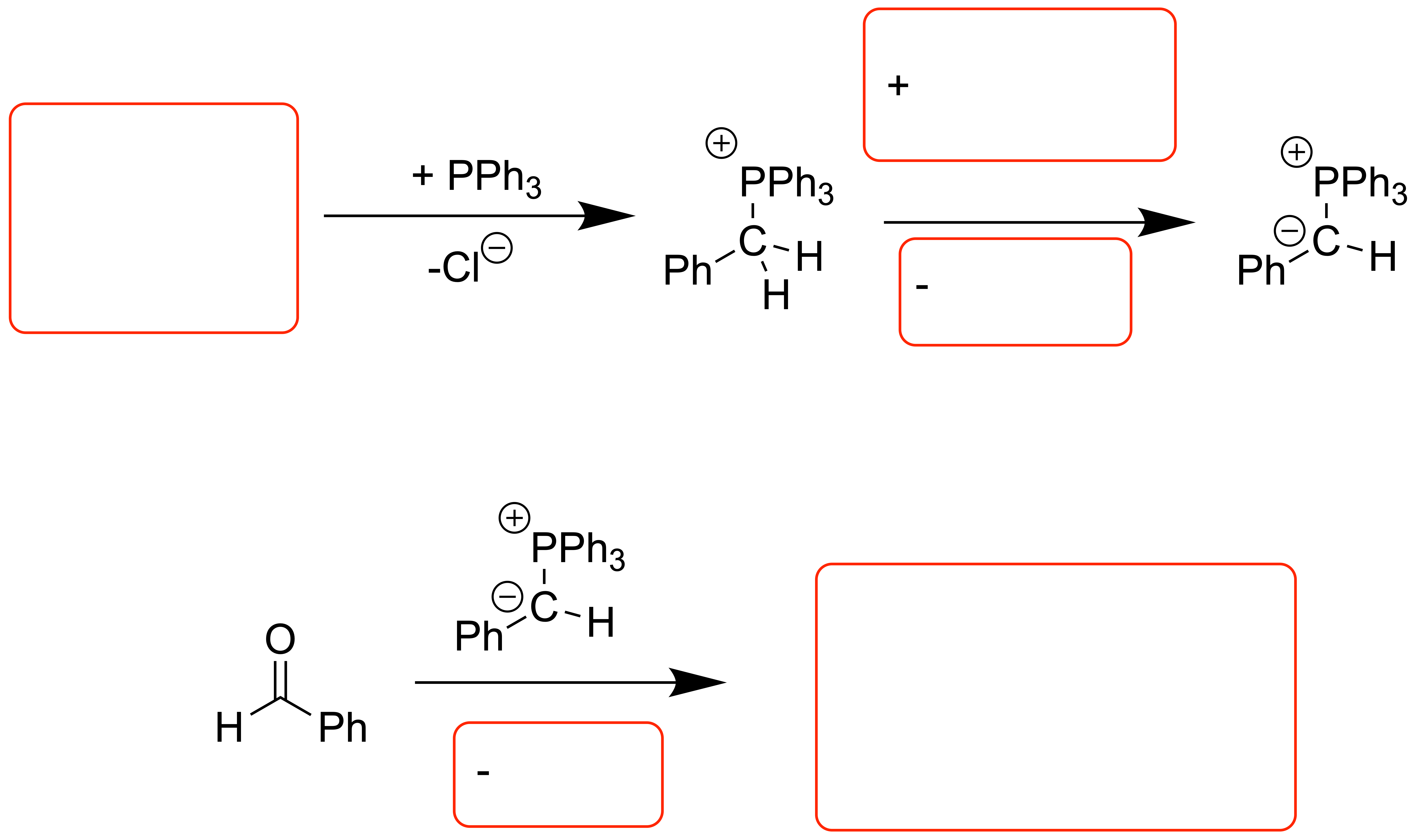
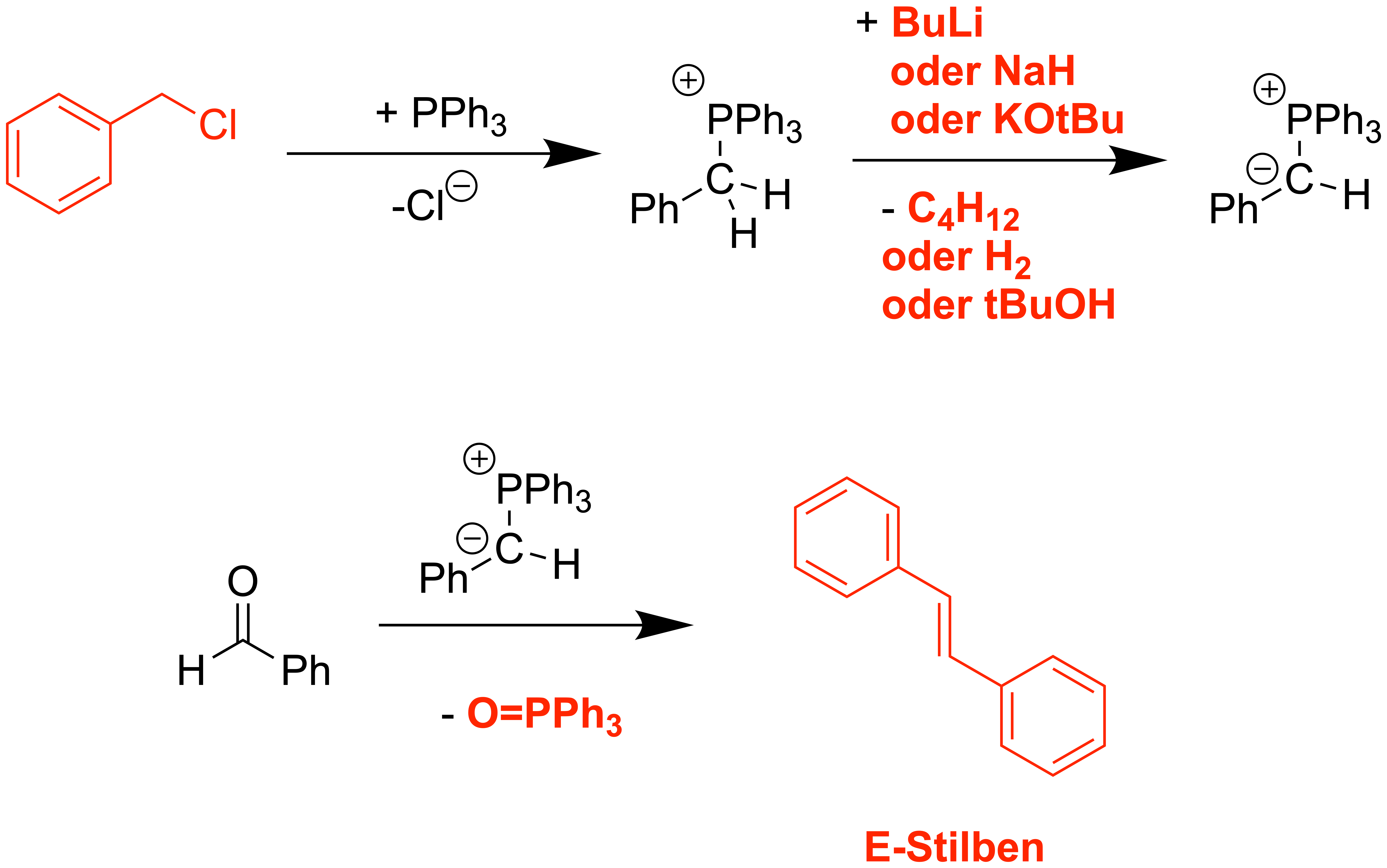
Das Ylid[1]


Ylide werden durch die Reaktion von Triphenylphosphan mit Methylchlorid (oder dessen Derivate, wenn andere Reste eingeführt werden sollen) dargestellt. Durch Deprotonierung des Substitutionsprodukts mit einer starken Base (hier Buthyl-Lithium) wird der einst elektrophile Kohlenstoff umgepolt – man erhält einen nucleophilen Kohlenstoff, gebunden an einen elektrophilen Phosphor.
Woher stammt der Begriff "Ylid"?
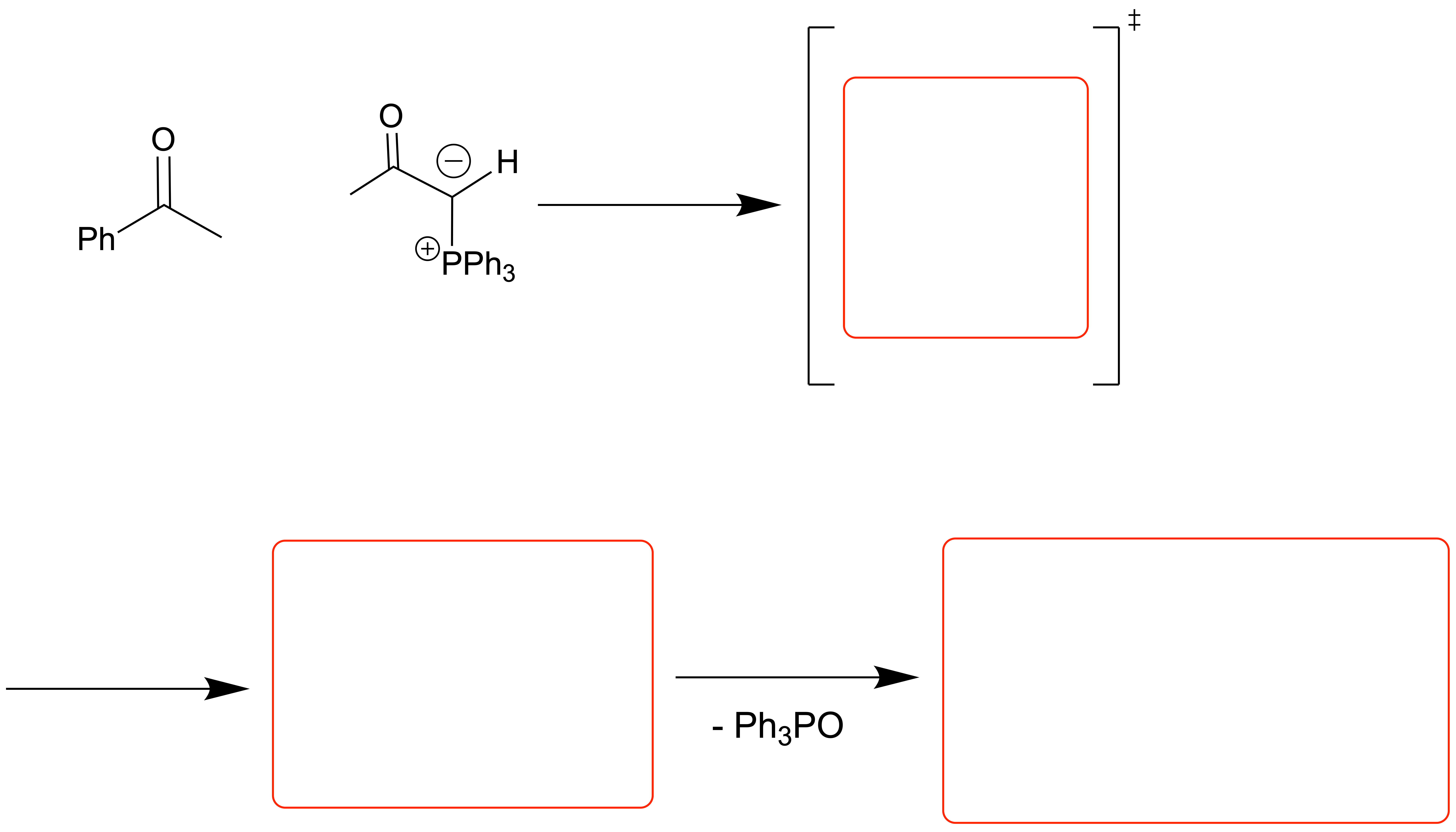
Das cyclische Interdukt wird (meistens) in einem Schritt gebildet[1], [2]


Achtung: Es besteht zwar mittlerweile Konsens über den hier aufgeührten ersten Reaktionsschritt, jedoch nur in Abwesenheit von Li+-Ionen, wovon auch im folgenden der Einfachheit halber ausgegangen wird. In vielen Lehrbüchern wird ein bereits widerlegter Mechanismus gezeigt, welcher über ein Betain-Intermediat verläuft.[2] Prüfe also zuvor, ob dies mit Deinem Lehrbuch im Einklang ist!
Der erste Schritt ist eine [2+2]-Cycloaddition und verläuft somit konzertiert. Es bildet sich ein Oxaphosphetan als Intermediat, welches sich durch eine syn-Eliminierung (oder Retro-Cycloaddition, wenn man so will) stabilisiert – es entsteht das Alken und ein Phosphanoxid-Derivat. Die P=O-Bindung ist sehr stark und eine wichtige Triebkraft dieser Reaktion. Dies ist wichtig, da diese Bindung das Gleichgewicht stark auf Seiten des Produkts dieses Reaktionswegs verschiebt.
Die S=O-Bindung ist beispielsweise nicht so stark, weshalb Schwefel-Ylide Dimethylsulfid(-Derivate) eliminieren, anstelle von Dimethylsulfoxid, und Oxirane anstelle von Alkenen bilden:

Die Art des Ylids ist maßgebend für die Stereoselektivität[1]
Da es sich bei dem Reaktionsprodukt um ein Alken handelt, unterscheidet man das E- und das Z-Produkt dieser Reaktion. Die Unterscheidung ist in erster Linie von Nöten, wenn das Ylid einen Substituenten am Kohlenstoff trägt; dieser hat dann einen maßgeblichen Einfluss auf die Stereoselektivität. Man unterscheidet bei der Wittig-Reaktion zwei Ylid-Klassen, welche mit beispielhaften Vertretern aufgeführt sind: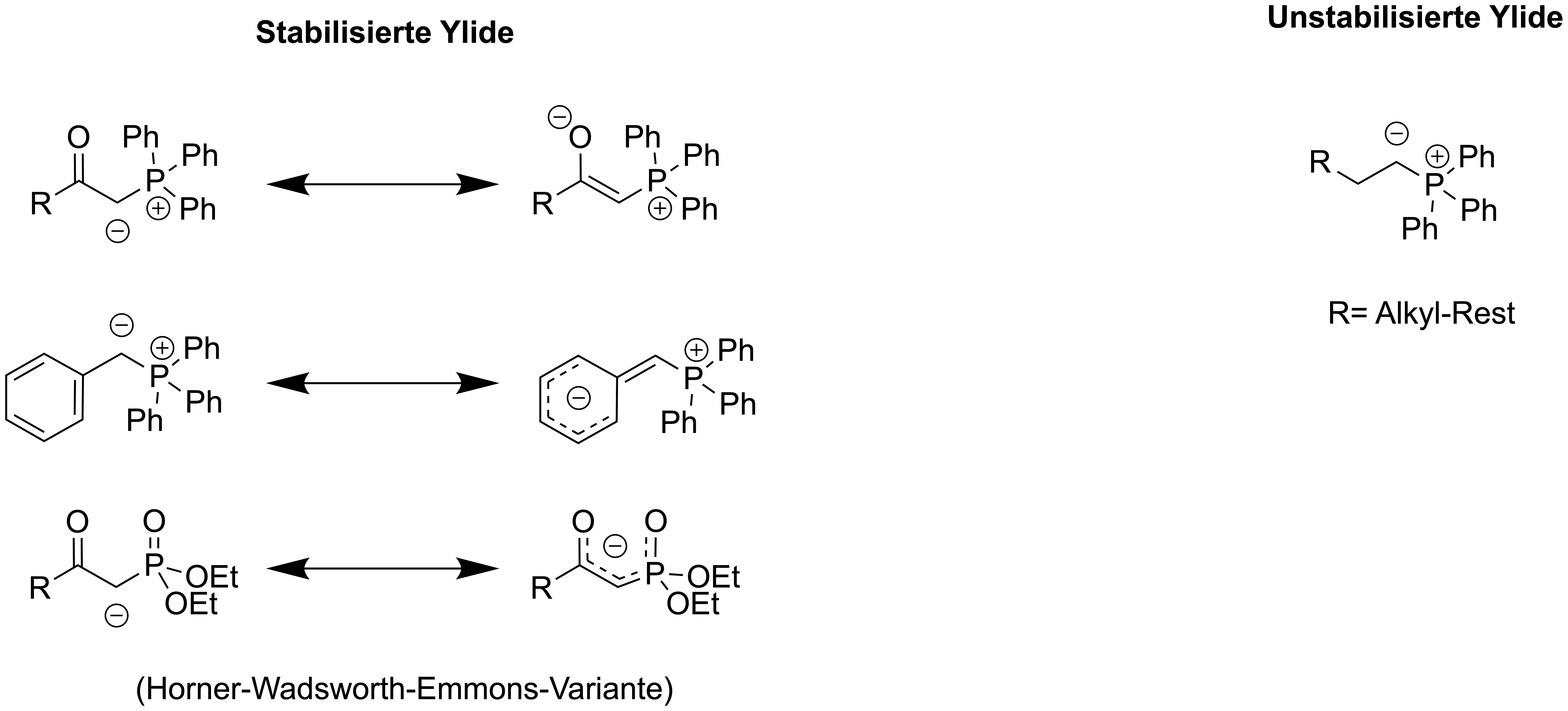
Unstabilisierte Ylide bilden das Z-Produkt[1]
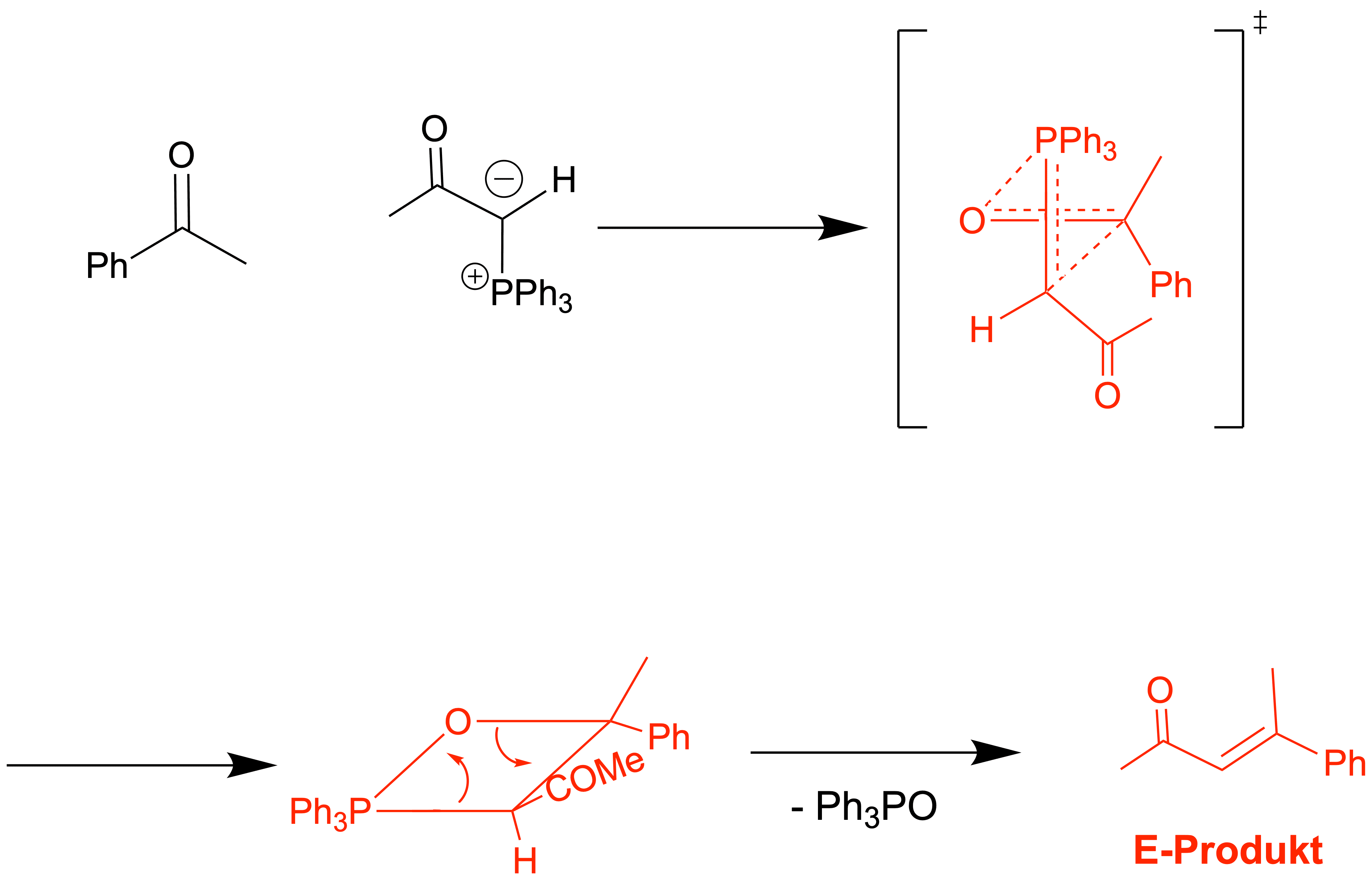
Da Z-Alkene meistens instabiler sind als E-Alkene, muss die Wittig-Reaktion unstabilisierter Ylide unter kinetischer Kontrolle liegen. Hierfür werfen wir einen Blick auf den Übergangszustand der Cycloaddition: Im Übergangszustand des geschwindigkeitsbestimmenden Schritts nähert sich das Ylid senkrecht an die Carbonyl-Funktion an
– hierbei ist jene Annäherung energetisch günstig, in welcher sich der Rest R' des Carbonyls möglichst weit weg von der PPh3-Gruppe und
dem Alkyl-Rest R des Ylids befindet. Wenn sich nun die beiden σ-Bindungen ausbilden, dreht sich das Ylid einmal um 90° (in diesem Fall gegen den Uhrzeigersinn): die beiden
Reste befinden sich nun auf der selben Seite und stehen folglich nach Eliminierung „Z“ konfiguriert zueinander – das thermodynamisch
ungünstigere Produkt wird selektiv gebildet!
Im Übergangszustand des geschwindigkeitsbestimmenden Schritts nähert sich das Ylid senkrecht an die Carbonyl-Funktion an
– hierbei ist jene Annäherung energetisch günstig, in welcher sich der Rest R' des Carbonyls möglichst weit weg von der PPh3-Gruppe und
dem Alkyl-Rest R des Ylids befindet. Wenn sich nun die beiden σ-Bindungen ausbilden, dreht sich das Ylid einmal um 90° (in diesem Fall gegen den Uhrzeigersinn): die beiden
Reste befinden sich nun auf der selben Seite und stehen folglich nach Eliminierung „Z“ konfiguriert zueinander – das thermodynamisch
ungünstigere Produkt wird selektiv gebildet!
Exkurs: Warum nähern sich Ylid und Carbonyl im rechten Winkel aneinander an?

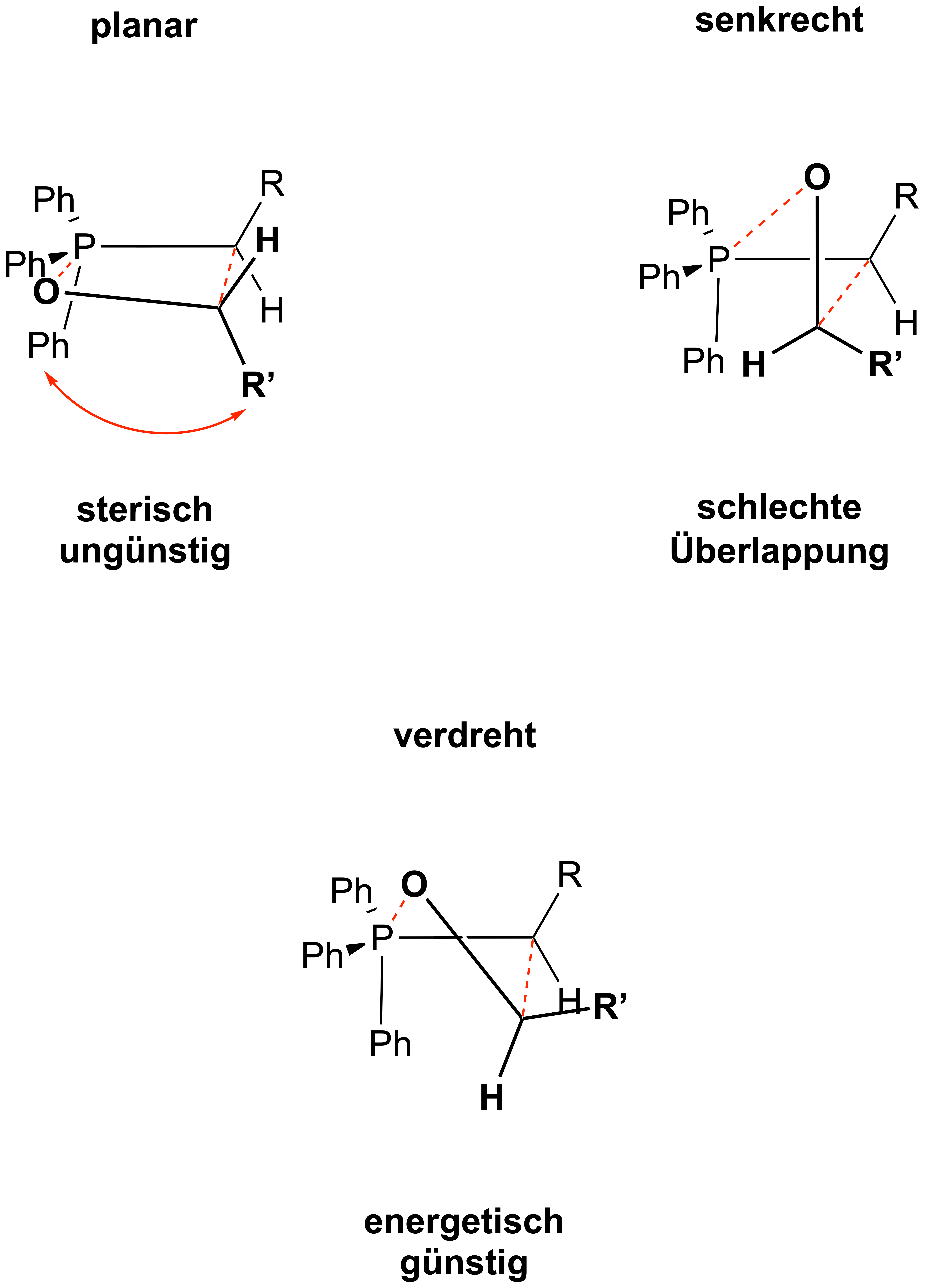 Es handelt sich also eher um eine Art verdrehten (engl.: puckered) Übergangszustand.
Es handelt sich also eher um eine Art verdrehten (engl.: puckered) Übergangszustand.
Der Grund liegt in der stark abstoßenden sterischen Wechselwirkung zwischen der Phenyl-Gruppe des Ylids und des Substituenten R' des Carbonyls. Durch die tetraedrische Geometrie des Phosphors sind dessen Phenyl-Substituenten so angeordnet, dass ein Rest anti-periplanar zum Substituenten R des Ylids steht, da dies die stabilste Konformation ist. Diese Phenyl-Gruppe schaut hier im Bild nach unten und kommt so in Konflikt mit dem Rest R' des Carbonyls, was einen planaren Übergangszustand energetisch stark ungünstig macht. Die verdrehte Anordnung sorgt somit für die minimalste sterische Wechselwirkung. Die Bergründung für die Z-Selektivität bleibt dieselbe, wie oben erläutert.
So kann man auch die E-Selektivität von Yliden erklären, welche sterisch weniger anspruchsvoll sind bezüglich des Phosphors (z.B. bei einer PPh2Me-Gruppe am Ylid).
Da herausgefunden wurde, dass die Cycloaddition in den meisten Fällen (und im Li+-freien Milieau) irreversibel ist, würde die unten aufgeführte Begründung für die E-Selektivität bei stabilisierten Yliden häufig nicht greifen, da sie von einer reversiblen Cycloaddition ausgeht. Die Ursache liegt in dem Fall in einer veränderten Geometrie des Phosphors im Übergangszustand: Bei stabiliserten Yliden wird ein später Übergangszustand postuliert (vgl. Hammond-Postulat). Dies bedeutet vorallem, dass die Umhybdridisierung im Übergangszustand schon weit fortgeschritten ist, wodurch der Phosphor nun eine trigonal bipyramidale Struktur besitzt. Dies ermöglicht einen nahezu planaren Übergangszustand, welcher zum E-Alken führt, ohne dass es zur oben genannten sterischen Wechselwirkung kommt. Nun ist auch der E-bildende Übergangszustand dem Z-bildenden bevorzugt.[2]
 An dieser Stelle will ich auf die Review von P. Byrne und G. Gilheany verweisen, auf die ich mich hier beziehe. Es ist zwar sehr lang und zäh geschrieben, besitzt allerdings einen unglaublich hohen Informationsgehalt und bringt endlich Licht ins Dunkle, was der aktuelle Stand der Dinge ist bei der Forschung rund um die Wittig-Reaktion.
An dieser Stelle will ich auf die Review von P. Byrne und G. Gilheany verweisen, auf die ich mich hier beziehe. Es ist zwar sehr lang und zäh geschrieben, besitzt allerdings einen unglaublich hohen Informationsgehalt und bringt endlich Licht ins Dunkle, was der aktuelle Stand der Dinge ist bei der Forschung rund um die Wittig-Reaktion.
Stabilisierte Ylide bilden das E-Produkt[1]
Warum das E-Alken bei stabilisierten Ylide gebildet wird, hängt hier auch wieder vom Ylid selber ab. Man kann aber allgemein zwei häufige Ursachen nennen:1. Akzeptor-Akzeptor-Repulsion
Dies betrifft hauptsächliche Ylide, welche selbst durch Carbonylgruppen stabilisiert werden; diese verändert nämlich nun die Anordnung im Übergangszustand: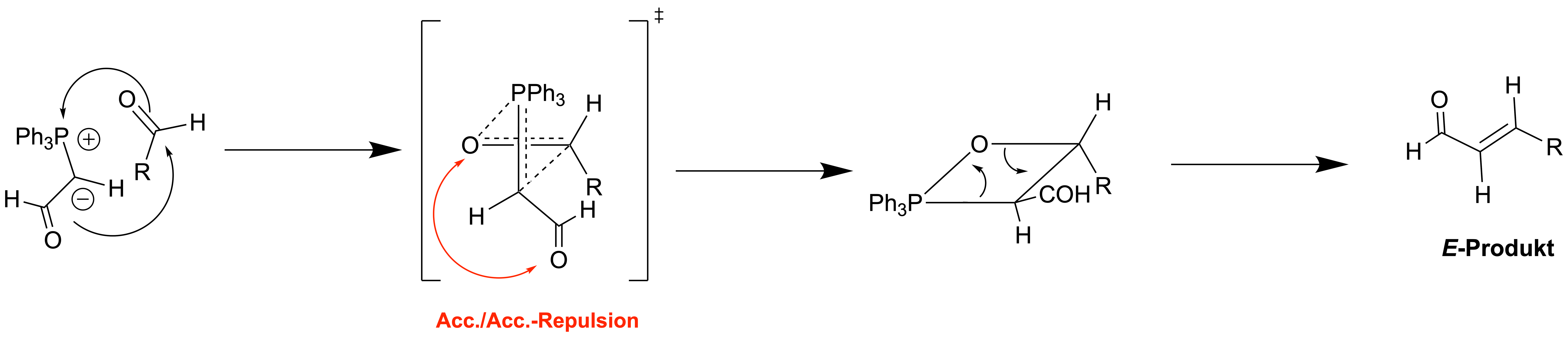
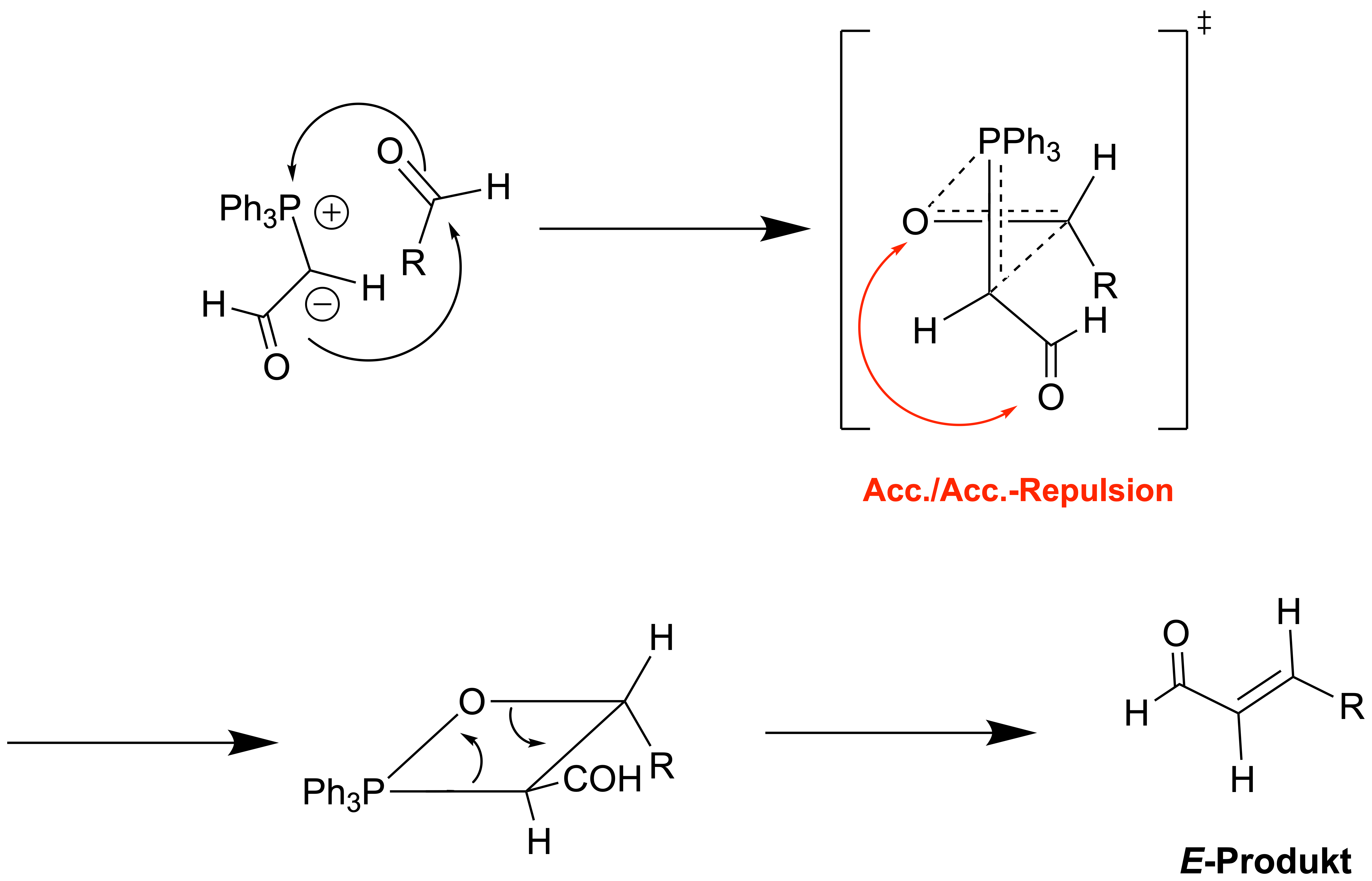
Die beiden elektronegativen Sauerstoffe der Carbonyl-Gruppen stoßen sich aufgrund der Coloumb-Kräfte stark ab; dieser Effekt spielt im Übergangszustand eine wichtigere Rolle als die sterische Abstoßung zwischen dem Rest R und der Carbonyl-Funktion. Somit ändert sich die Kinetik und das E-Produkt wird selektiv gebildet.
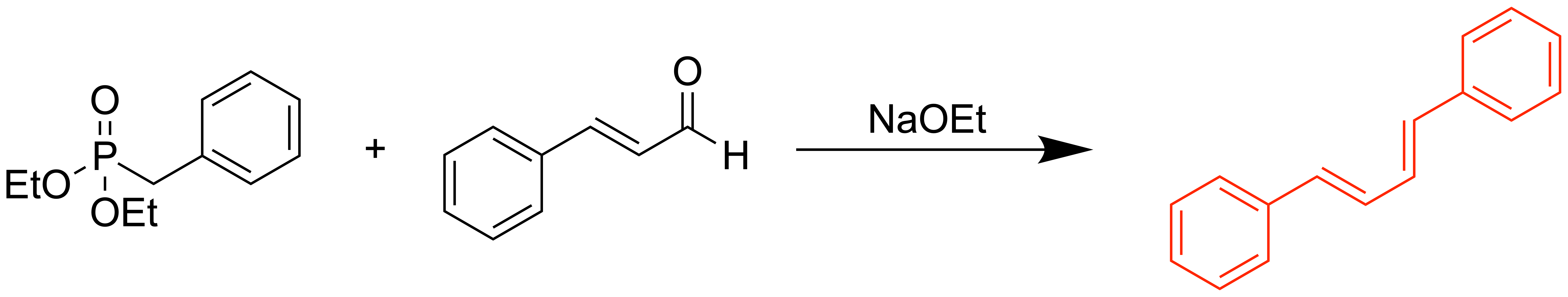
2. Reversibilität der Interduktbildung
Dies betrifft zum Beispiel die sogenannten Horner-Ylide, wie bei der Horner-Wadsworth-Emmons-Reaktion: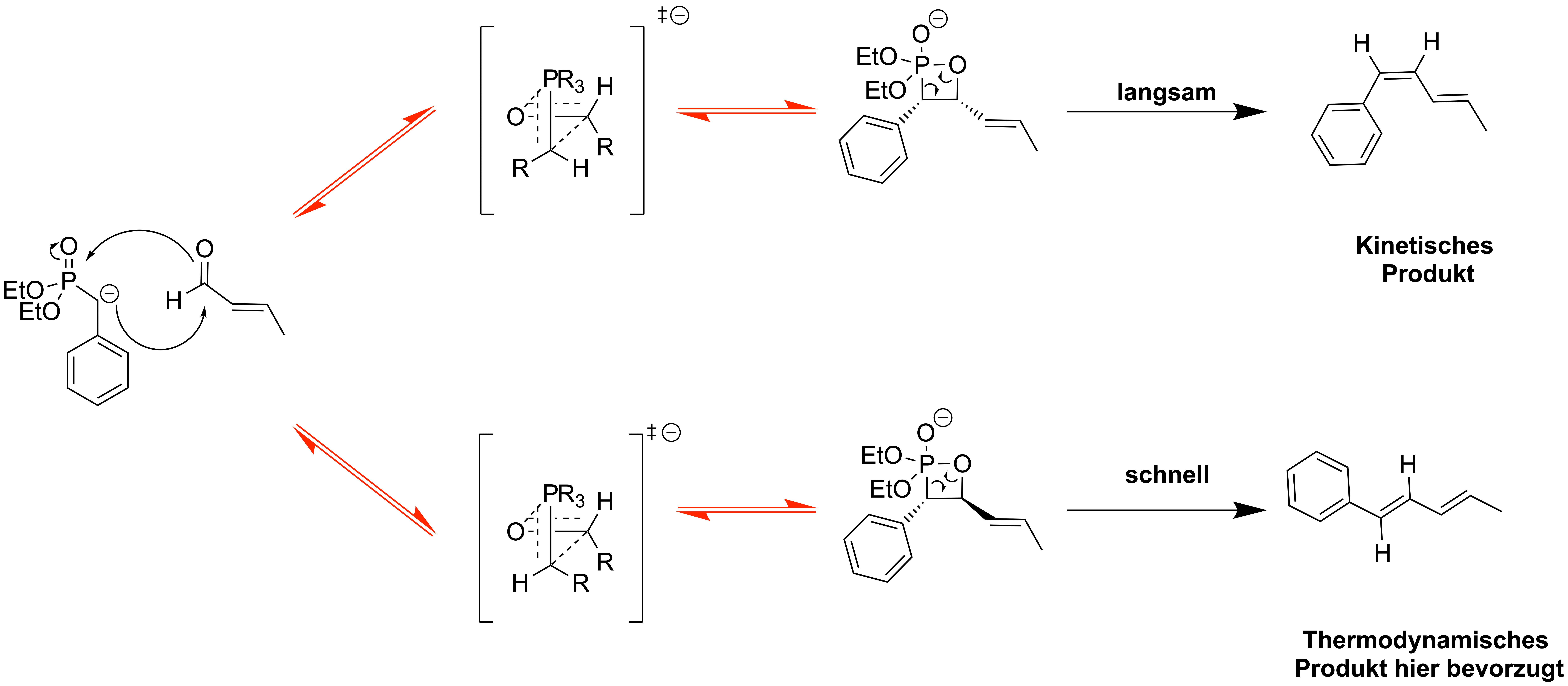
Horner-Ylide sind durch die starke Delokalisation besonders stabilisiert, und sorgen dafür, dass die Bildung des cyclischen Interdukts nun reversibel ist. Wenn sich jetzt das Interdukt bildet, welches zum Z-Alken führt, profitiert man jetzt davon, dass die syn-Eliminierung sehr langsam verläuft (bedeutet hohe Aktivierungsenergie). Somit gibt es genug Zeit für das Intermediat um zurückzureagieren.
Die Moleküle, welche sich jedoch auf dem anderen Weg mit höherem sterischen Anspruch und höherer Aktivierungsenergie annähern und das untenstehende Intermediat bilden, reagieren daraufhin schnell zum E-Alken. Die Aktivierungsenergie ist hier niedriger als bei der Eliminierung zum Z-Alken. Das Intermediat hat also weniger Zeit um zurückzureagierem, weshalb somit insgesamt das E-Alken bevorzugt gebildet wird. Die Reaktion liegt nun unter thermodynamischer Kontrolle.
Warum passiert das nicht bei unstabilisierten Yliden?
Bei Horner-Yliden ist die Energiedifferenz zum Intermediat nicht mehr so hoch, da das Ylid energetisch abgesenkt wurde. Die Rückreaktion findet nun im bemerkbaren Ausmaß statt.
Auf einem Blick...
Z-Produkt
• Unstabilisierte Ylide
• Kinetische Kontrolle
• Substituenten stoßen sich im ÜZ ab

E-Produkt
• Stabilisierte Ylide
• Thermodynamische Kontrolle
• Acc./Acc.-Repulsion und/ oder reversible Interduktbildung
• HWE-Reaktion mit Horner-Yliden
Referenzen
1. J.Clayden, N.Greeves, S.Warren, P.Wothers in Organic Chemistry, Vol. 8, Oxford University Press, Oxford, 2008.
2. P.Byrne, D.Gilheany, Chem. Soc. Rev. 2013, 42, 6670-6696.
Wenn nicht anders angegeben, sind alle Abbildungen selbst angefertigt.