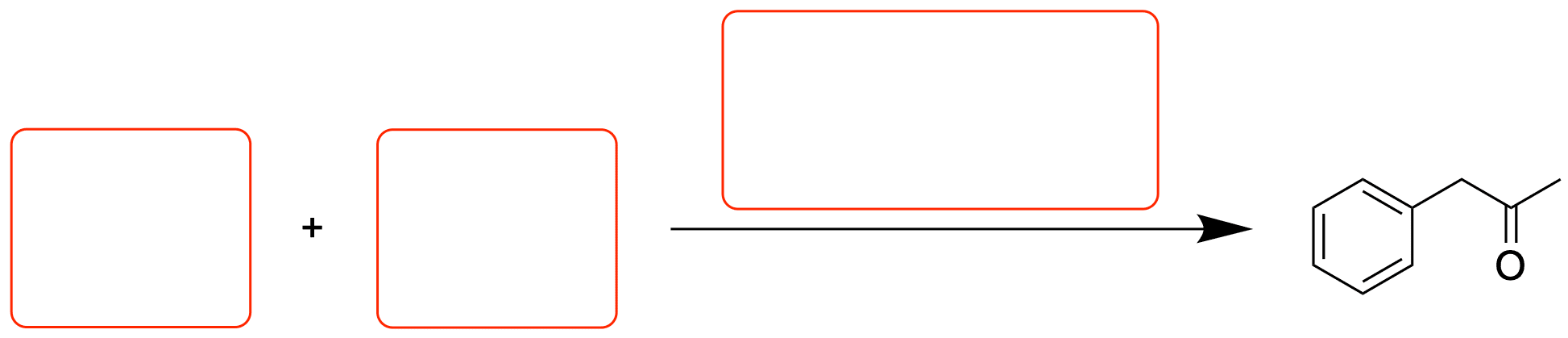
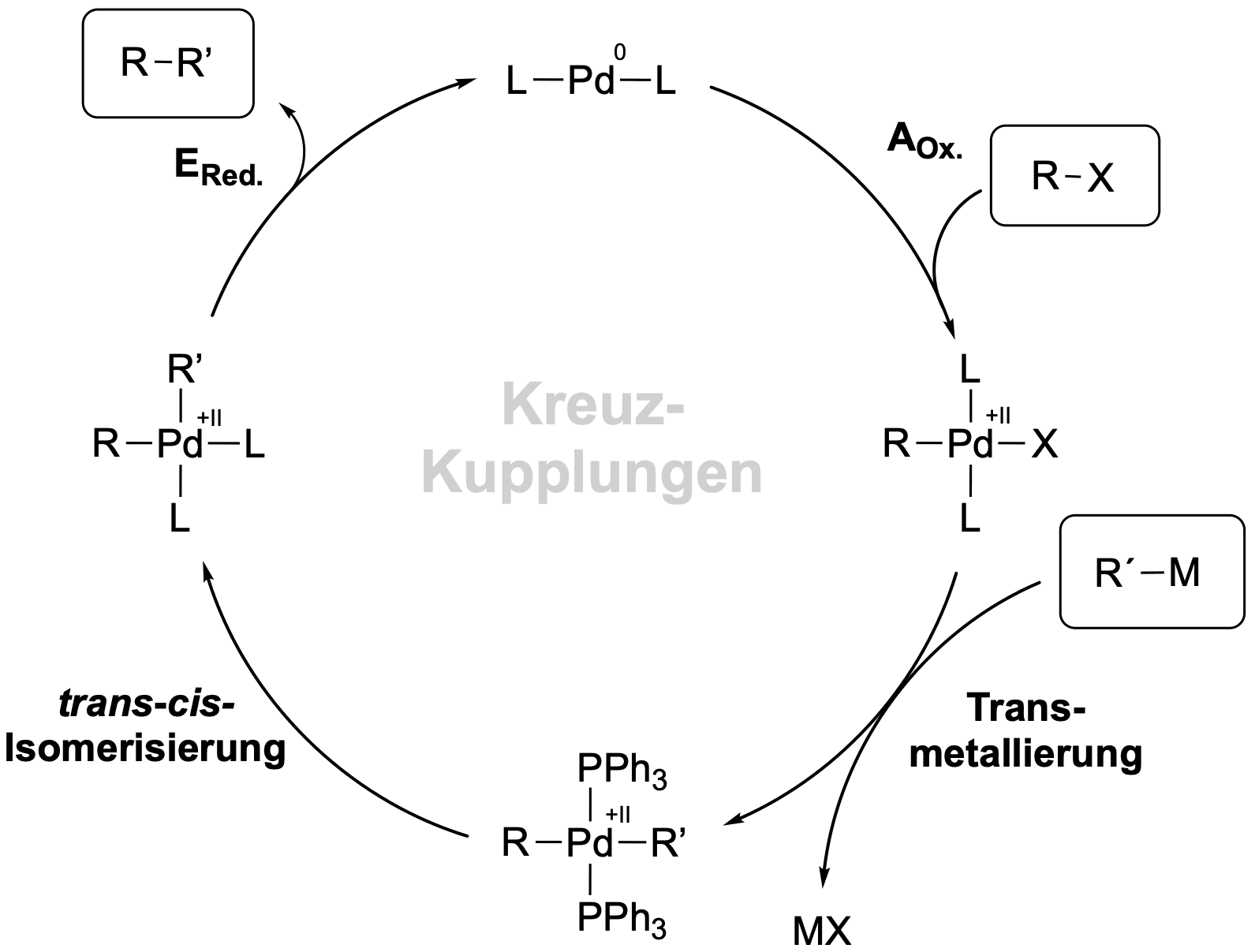
 Kreuzkupplungen sind aus der modernen Synthesechemie nicht mehr wegzudenken. Doch wer zum ersten mal das Schema einer Kreuzkupplung gesehen hat, wird dies vermutlich nicht direkt erkennen. Schauen wir uns ein solches Schema mal an:
Kreuzkupplungen sind aus der modernen Synthesechemie nicht mehr wegzudenken. Doch wer zum ersten mal das Schema einer Kreuzkupplung gesehen hat, wird dies vermutlich nicht direkt erkennen. Schauen wir uns ein solches Schema mal an:
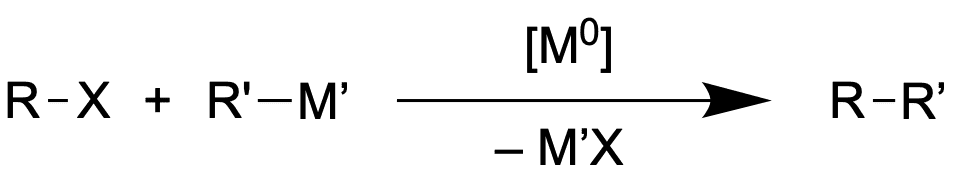
Die Komponente R-X setzt sich zusammen aus dem organischen Rest R und der kohlenstoffgebundenen Abgangsgruppe X. Hierbei kann es sich um ein Halogenid handeln (Bromid, Iodid), aber auch um ein Triflat (-OSO2CF3). Allgemein gesprochen liegt uns hier ein Elektrophil vor, da der X-gebundene Kohlenstoff eher positiv teilgeladen ist.
Der Kupplungspartner R-M stellt hingegen das Nucleophil dar, da es sich hierbei um eine Organometall-Verbindung handelt und der M-gebundene Kohlenstoff somit negativ (teil-)geladen ist. Bei dem Metall M kann es sich um verschiedene Metalle handeln: So können Lithium-Organyle, Grignard-Verbindungen, Cuprate oder sogar Boronate eingesetzt werden, wie wir noch sehen werden.
Wir halten schonmal fest, dass es sich bei Kreuzkupplungsreaktionen um C-C-Bindungsknüpfungen handelt ( - Es gibt zwar auch Ausnahmen wie die Buchwald-Hartwig-Kupplung, aber dazu später). Wie genau die Reste R aussehen definieren wir zunächst nicht.
Die Reaktion dieser beider Komponenten findet aber nicht ohne weiteres statt:

Eine Reaktion dieser Art ist bspw. die Wurtz-Fittig-Reaktion. Diese Reaktionen funktionieren nur in besonderen Fällen und liefern häufig schlechte Ausbeuten und viele Nebenprodukte. Anders sieht es aus, wenn wir einen Katalysator einsetzen. Hierbei handelt es sich um ein Übergangsmetall in der Oxidationsstufe 0. Am weitesten verbreitet ist der Einsatz von Palladium, worauf wir uns auch im Folgenden beschränken werden, es ist aber auch möglich Nickel, Kupfer oder gar Eisen als ökonomische Variante einzusetzen.
Das Palladium koordiniert als aktive Katalysatorspezies noch zwei Liganden L, wobei hier häufig Triphenylphosphan-Liganden (PPh3) eingesetzt werden. Somit sähe eine mögliche Reaktionsgleichung wie folgt aus:
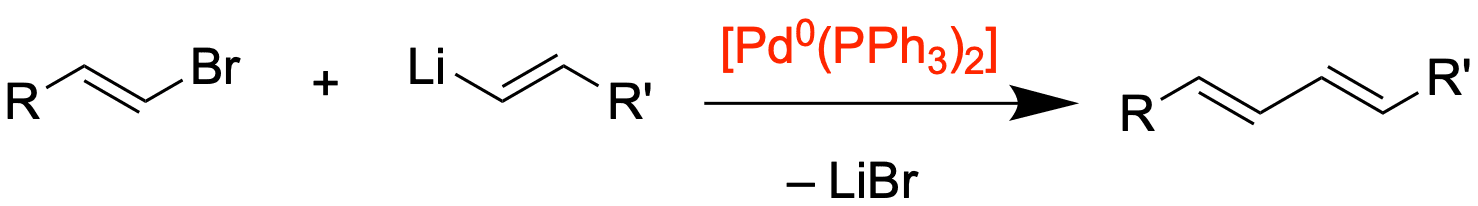
Der Reaktionsmechanismus verläuft dementsprechend auch nicht trivial über einen nucleophilen Angriff, sondern über einen Katalysezyklus, bei welcher vier wesentliche Reaktionen durchlaufen werden. Zuvor schauen wir uns aber Palladium nochmal genauer an.
Palladium und seine zwei wichtigen Oxidationsstufen[1]
Palladium ist ein Übergangsmetall aus der 10. Gruppe und besitzt somit 10 Valenzelektronen in der Oxidationsstufe 0. In der wichtigen Oxidationsstufe +II verfügt es über 8 Valenzelektronen. Da dort ein d8-System vorliegt, koordiniert es quadratisch planar, wie z.B. in diesem Komplex: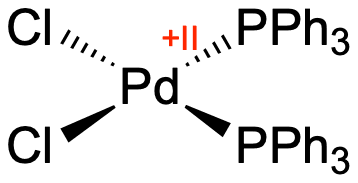
Wohlgemerkt handelt es sich bei den Triphenylphosphan-Liganden (PPh3) rechts um neutrale Liganden.
In der Oxidationsstufe 0 bindet es ebenfalls vier Liganden, da es so die 18-Elektronen-Regel erfüllt. Ein Beispiel ist Tetrakis(triphenylphosphan)palladium(0):
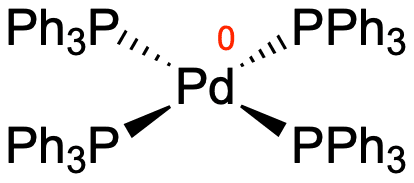
Dieser Komplex ist stabil und könnte uns somit nicht als Katalysator dienen. Es kann in Lösung jedoch zu einer Dissoziation zweier Liganden kommen, wodurch ein sehr reaktiver Komplex entsteht, da er koordinativ ungesättigt ist. Das heißt, es verfügt über freie Bindestellen für Liganden und kann somit leicht Reaktionen eingehen.
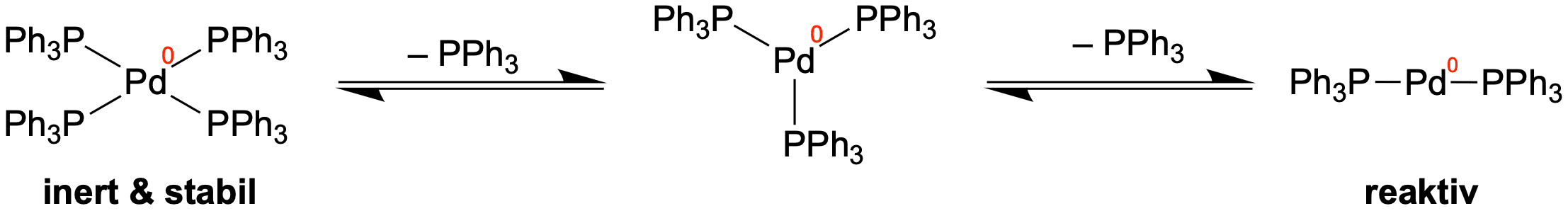
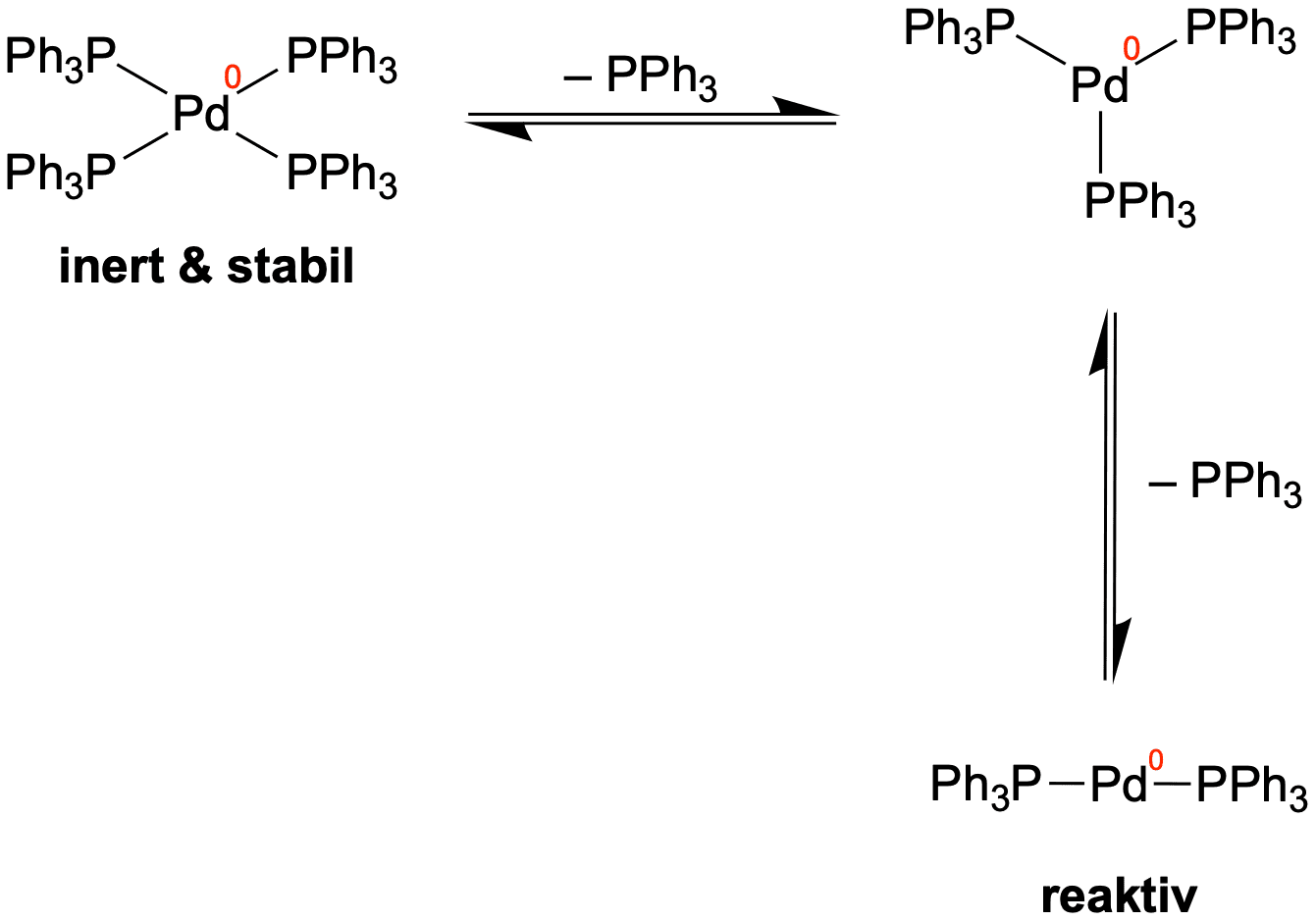
Von diesem linear koordinierten Komplex aus startet auch die Kreuzkupplung. Wir sollten aber im Kopf behalten, dass in Lösung hauptsächlich der Palladium-Komplex mit vier Liganden vorliegt. Wenn Reaktionsgleichungen oder Katalaysezyklen angegeben werden, wird der Schritt der Dissoziation jedoch häufig weggelassen.
Wie wir sehen werden, wechselt Palladium während der Reaktion zwischen diesen beiden Oxidationsstufen und ermöglicht so die Kreuzkupplung.[1]
Der Katalysezyklus setzt sich aus vier Reaktionen zusammen[1]
Wir schauen uns nun einen konkreten Katalysezyklus an und gehen die einzelnen Reaktionsschritte nacheinander durch.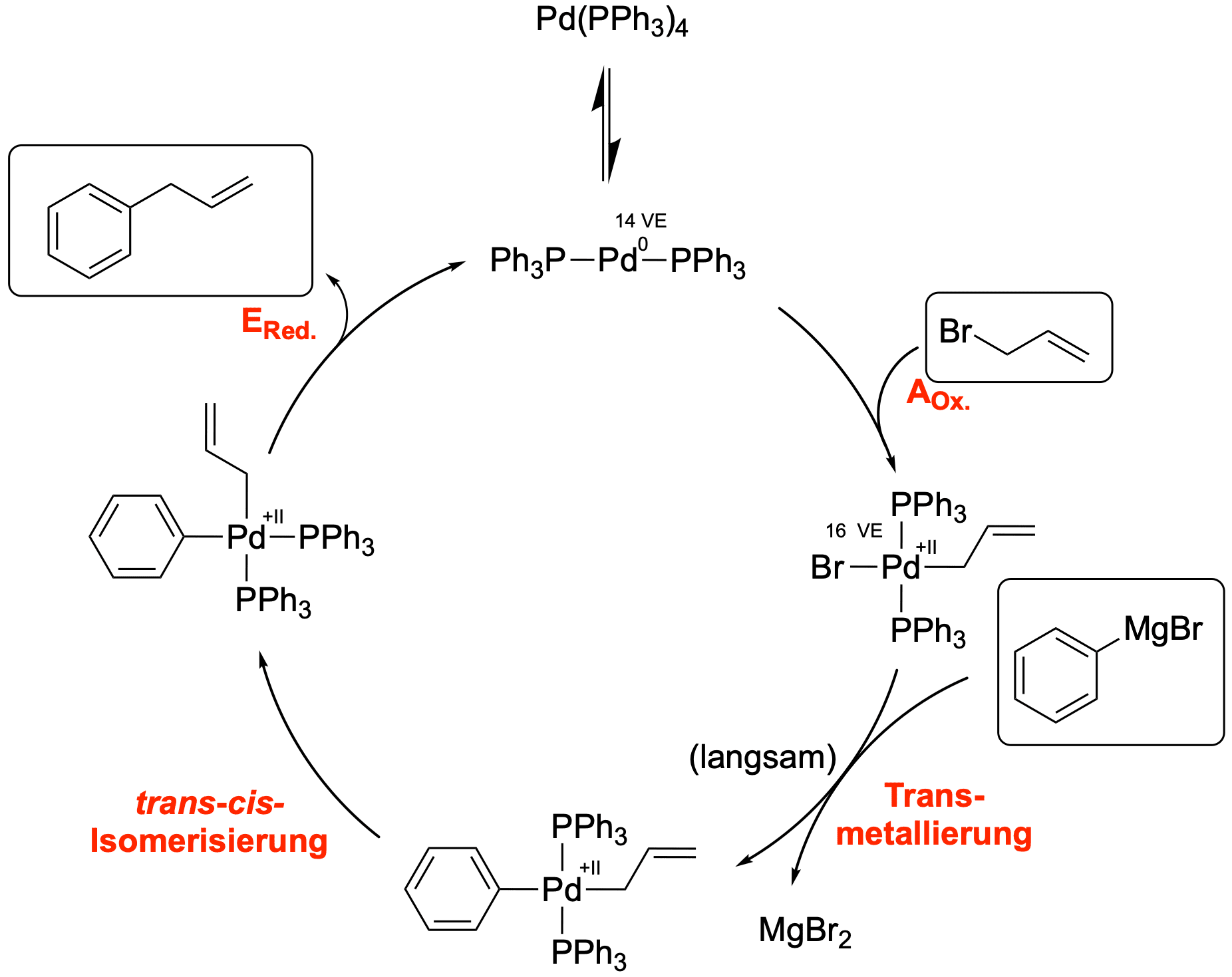
Die Oxidative Addition AOx
Die Oxidative Addition ist der erste Schritt des Katalysezyklus und stellt eine Reaktion zwischen dem linearen Palladium(0)-Komplex und dem Elektrophil dar.
Das Palladium sättigt seine Koordinationssphäre indem es sich in die C-X-sigma-Bindung (hier die C-Br-Bindung) insertiert; vergleichbar mit der Reaktion zur Darstellung einer Grignard-Verbindung (R-Br + Mg → R-MgBr). Der Kohlenstoff des Restes R wird hierbei umgepolt, da es reduziert wurde und deshalb nach der Addition als Carbanion vorliegt. Im Gegenzug wurde das Palladium auf die Oxidationsstufe +II oxidiert.
Wodurch wird diese Reaktion begünstigt?
Die Transmetallierung
Das vierfach koordinierende Intermediat reagiert im nächsten Schritt mit der Organometall-Verbindung. Dieser Schritt ist der langsamste Schritt dieses Zyklus und somit der geschwindigkeitsbestimmende Schritt.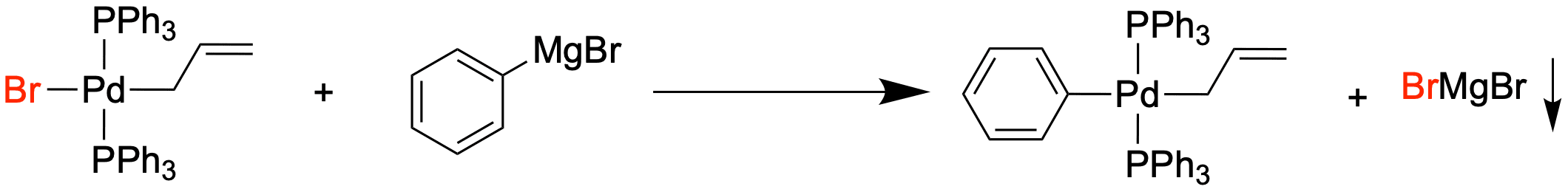
Bei der Transmetallierung tauschen zwei Liganden das Metall, an denen sie koordiniert sind. Das Carbanion wechselt vom Magnesium zum Palladium, während das Bromid in unserem Fall zum Magnesium wechselt - die Transmetallierung kann demnach auch als Metathese aufgefasst werden. Wenn Magnesiumbromid ausfallen kann, bringt dies auch noch eine starke Triebkraft für die Kupplung mit sich.
Die trans-cis-Isomerisierung
Die folgenden Reaktionen sind beide intramolekular und dementsprechend schnell. Damit das Reaktionsprodukt erhalten werden kann, muss eine Eliminierung stattfinden. Da die Eliminierung als konzertierter Schritt abläuft, ist es notwendig, dass die beiden organischen Reste cis zueinander stehen. Da trans-Komplexe ohnehin schon instabiler sind, als cis-Komplexe (trans-Einfluss), geht die Isomerisierung sehr bereitwillig von statten.
Die reduktive Eliminierung ERed
Wie schon erwähnt wird bei der reduktiven (syn-) Eliminierung zum einen das Produkt eliminiert, zum anderen erhalten wir den linearen Pd(0)-Komplex wieder, welcher wieder neue reagieren kann.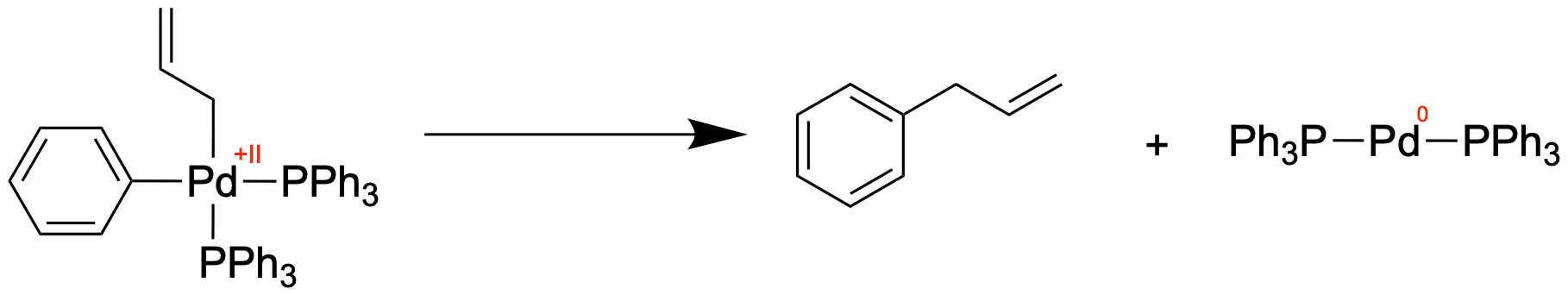
Ist die reduktive Eliminierung die Rückreaktion der oxidativen Addition?
Wenn jedoch die ERed im letzen Schritt des Katalysezyklus mit der AOx des ersten Schritts verglichen werden soll, so kann es mechanistische Unterschiede geben, da wir verschiedene Substrate betrachten. In diesem Fall wäre es streng genommen ungenau, diese beiden Reaktionen mechanistisch als gleich anzusehen.
Der Einsatz von Katalyse-Precursoren[1]
Ein weiterer wichtiger Aspekt moderner Kreuzkupplungen ist, dass häufig kein Pd(0)-Komplex in die Reaktionslösung gegeben wird, sondern eine Pd(II)-Spezies, da Pd(0)-Komplexe nicht luftstabil sind. So wird in der Literatur häufig solch eine Reaktion angegeben:
Der Grund, warum die Reaktion trotzdem funktioniert, ist, dass die Pd(0)-Spezies in situ dargestellt wird. Wir geben also eine Vorläufer-Verbindung hinzu, welche mit Triethylamin (TEA bzw. NEt3) reagiert und so die aktive Katalysatorspezies liefert:
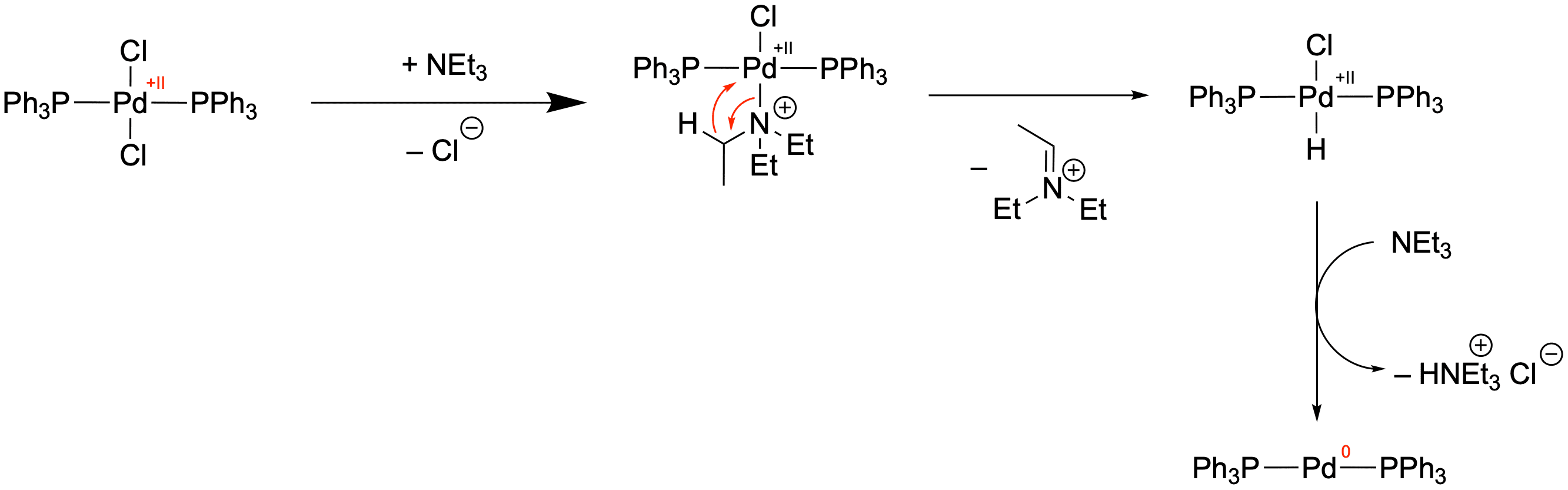
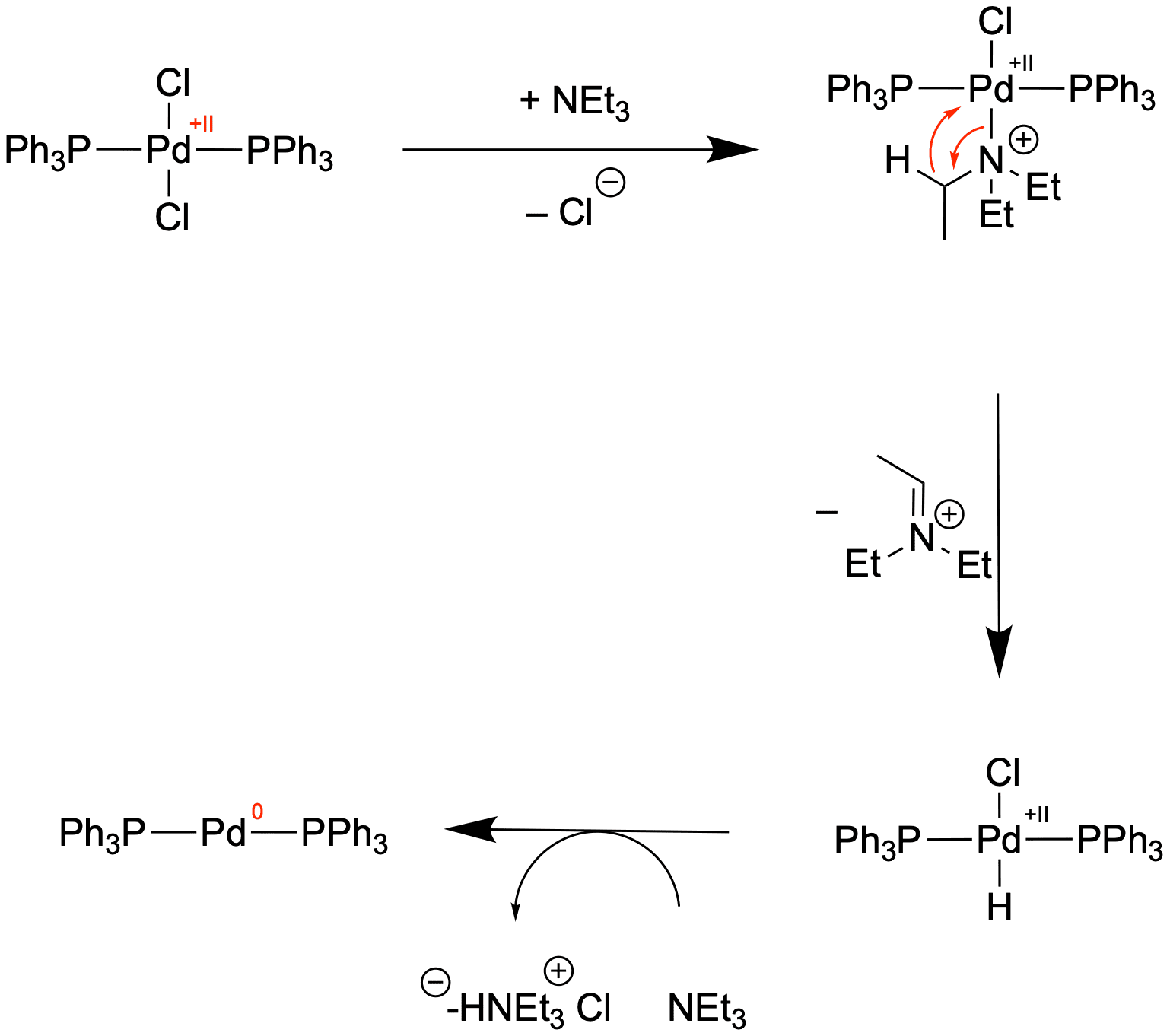
In dieser Reaktionssequenz wird das Palladium durch Triethylamin reduziert, welches selbst zu einem Iminium-Ion oxidiert wird. Diese Reduktion von Pd(0) setzt sich aus drei grundlegenden Reaktionen zusammen:
- Ligandenaustausch-Reaktion
Hier wird der Chlorido-Ligand durch einen Amin-Liganden ausgetauscht. - β-Hydrid-Eliminierung
Die β-H-Eliminierung ist eine wichtige Reaktion in der organometallischen Chemie und wird uns später nochmal wiederbegegnen. Hier wandert formal ein Hydrid von einem Kohlenstoff in β-Position zum Palladium (Stickstoff läge in der α-Position) zum Palladium als neuer Ligand. Gleichzeitig bildet sich eine π-Bindung aus und das Iminium-Ion wird eliminiert. - Reduktive Eliminierung
Diesen Schritt haben wir bereits kennengelernt. Nach der β-H-Eliminierung kann sich leicht HCl abspalten und unsere aktive Katalysator-Spezies wird dargestellt.
Kreuzkupplungen unterscheiden sich durch das Nucleophil[1]
Da wir jetzt alle Grundprinzipien haben, schauen wir uns nun vier konkrete Kreuzkupplungen an. Im Endeffekt machen sie alle dasselbe, aber es gibt einen wichtigen Faktor, der sie voneinander unterscheidet: Die Toleranz gegenüber funktionellen Gruppen.Wir fangen gleich mit der Reaktion an, welche nur wenige funktionelle Gruppen toleriert und enden mit einer Kreuzkupplung, welche so gut wie jede andere funktionelle Gruppe toleriert. Der Trend kommt daher, weil sich diese vier Reaktionen nur durch das Nucleophil (R-M) unterscheiden, welche sie verwenden. Genau genommen nimmt die Elektronegativitätsdifferenz zwischen dem Kohlenstoff und M immer weiter ab, sodass der kovalente Chrakakter dieser Bindung immer weiter zunimmt. Darus folgt auch eine Abnahme der Reaktivität, welche dafür sorgt, dass mehr funktionelle Gruppen toleriert werden.
Die Kumada-Kreuzkupplung
Die Kumada-Kupplung haben wir im Prinzip schon kennengelernt. Hier werden Grignard-Verbindungen (R-MgBr) als Nucleophile eingesetzt.
Die Substrate sind präparativ leicht zugänglich, da zur Darstellung des Nucleophils lediglich ein organisches Halogenid mit Magnesium in THF umgesetzt werden muss. Die größte Limitation jedoch liegt in der geringen Toleranz der Grignard-Verbindung gegenüber anderen funktionellen Gruppen. Dies betrifft vor allem Carbonyl-Funktionen und bereits leicht acide Gruppen wie Alkohole, Mercaptane oder Amine. Des Weiteren muss ebenfalls vollständig inert gearbeitet werden, da Grignard-Reagenzien stark hydrolyseempfindlich sind.
Die Negishi-Kreuzkupplung
Bei der Negishi-Kupplung werden Zink-Organyle eingesetzt, welche weniger Reaktiv als Grignard-Verbindungen sind.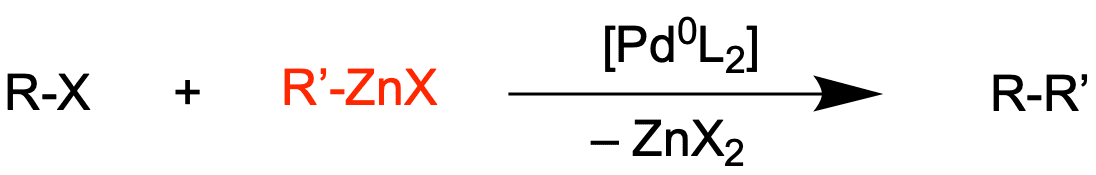
Organozink-Verbindungen sind ein ein wenig toleranter gegenüber funktionellen Gruppen. Außerdem sind sie reaktiver und weniger toxisch als Organozinn-Verbindungen, welche bei der Stille-Kupplung eingesetzt werden.
Stille-Kupplung
Bei der Stille-Kupplung wird ein Zinn-Organyl R‘-Sn(Bu)3 als Nucleophil eingesetzt. Dieses kann durch die Umsetzung des entsprechenden Metallorganyls mit Tributylzinnchlorid dargestellt werden, wobei es sich allgemein lediglich um einen Alkyl-Rest, und nicht zwingend um einen Butyl-Rest, handeln muss.
Die Stille-Kupplung findet große Anwendung in der Naturstoffsynthese, vor allem aufgrund der geringen Reaktivität des Zinnorganyls gegenüber anderen funktionellen Gruppen. So bedarf die Kupplung keiner Schutzgasatmosphäre und es kann in manchen Fällen sogar im wässrigen Milieu gearbeitet werden. Ein großer Nachteil liegt jedoch in der hohen Toxizität von Organozink-Verbindungen.
Suzuki-Kupplung
Mit wenig toxischen Substraten kommt die Suzuki-Kupplung aus. Hier werden Organoborane, Boronsäuren (R''=OH) oder Boronate (R''=OR) R‘–BR‘‘3 eingesetzt.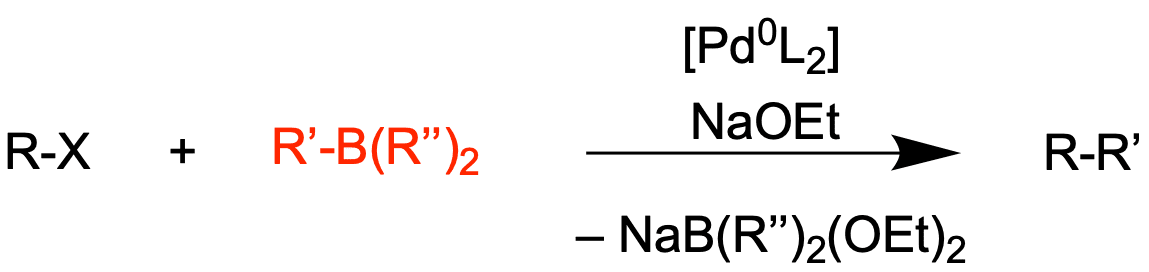
Die Reaktion kann im wässrigen Milieu durchgeführt werden. Da Boronsäuren bzw. Boronate wenig toxisch und sehr tolerant gegenüber anderen funktionellen Gruppen sind, ist die Suzuki-Kupplung vergleichsweise umweltfreundlich und im Allgemeinen sehr ökonomisch. Ein Nachteil liegt jedoch in den langen Reaktionszeiten von bis zu mehreren Stunden und hohen Temperaturen, welche mit der geringen Reaktivität des Nucleophils einhergehen.[2]
Wir halten fest: Besuche diese Seite auf einem Gerät mit größerem Bildschirm, um eine Übersicht zu erhalten!
| Kreuzkupplung | Nucleophil | Vorteile | Nachteile |
|---|---|---|---|
| Kumada-Kupplung | R-MgBr | • Nuc. leicht darzustellen
• Kurze Reaktionszeiten |
• Fehlende Toleranz gegenüber vielen funktionellen Gruppen
• Inertes Arbeiten notwendig |
| Negishi-Kupplung | R-ZnX | • Geringere Reaktivität als Grignard-Verbindungen
• Weniger toxisch als Zinn-Organyle |
• Geringe Toleranz gegenüber vielen funktionellen Gruppen
• Inertes Arbeiten notwendig |
| Stille-Kupplung | R-SnR3 | • Gute Toleranz gegenüber anderen funktionellen Gruppen
• Keine inerten Reaktionsbedingungen nötig • Manchmal kann im wässrigen Milieau gearbeitet werden |
• Hohe Toxizität von Organozinn-Verbindungen |
| Suzuki-Kupplung | R-BR2 | • Ausgezeichnete Toleranz gegenüber anderen funktionellen Gruppen
• Keine inerten Reaktionsbedingungen nötig • Durchführung im wässrigen Milieau möglich • Ökologisch & Ökonomisch |
• Ggf. längere Reaktionszeiten |
Grenzen der Kreuzkupplungen[1]
Mit Kreuzkupplungen sind viele Arten von Reaktionen zu realisieren. Allerdings gibt es eine wichtige Limitation: Für das Elektrophil kann nicht jeder beliebige Rest eingesetzt werden. So sind Kreuzkupplungen mit einem Alkyl-Rest am Elektrophil nicht realisierbar - wir werden aber noch eine genauere Bedingung formulieren.Der Grund für diese Limitation ist der Schritt der Transmetallierung. Sie ist, wie schon erwähnt, der langsamste Schritt dieser Reaktion. Unwissenschaftlich ausgedrückt, muss also das Pd(II)-Intermediat, welches sich nach der AOx bildet, einige Zeit "warten", bis es weiterreagieren kann. Hat dieses Intermediat die Möglichkeit, eine schnelle, intramolekulare Reaktion zu durchlaufen, so wird es hauptsächlich zu dieser Reaktion kommen, und nicht zur Transmetallierung. Diese konkurrierende Reaktion ist die schon diskutierte β-H-Eliminierung. Verwenden wir also bspw. 1-Brompropan als Elektrophil, würde hauptsächlich folgendes passieren:
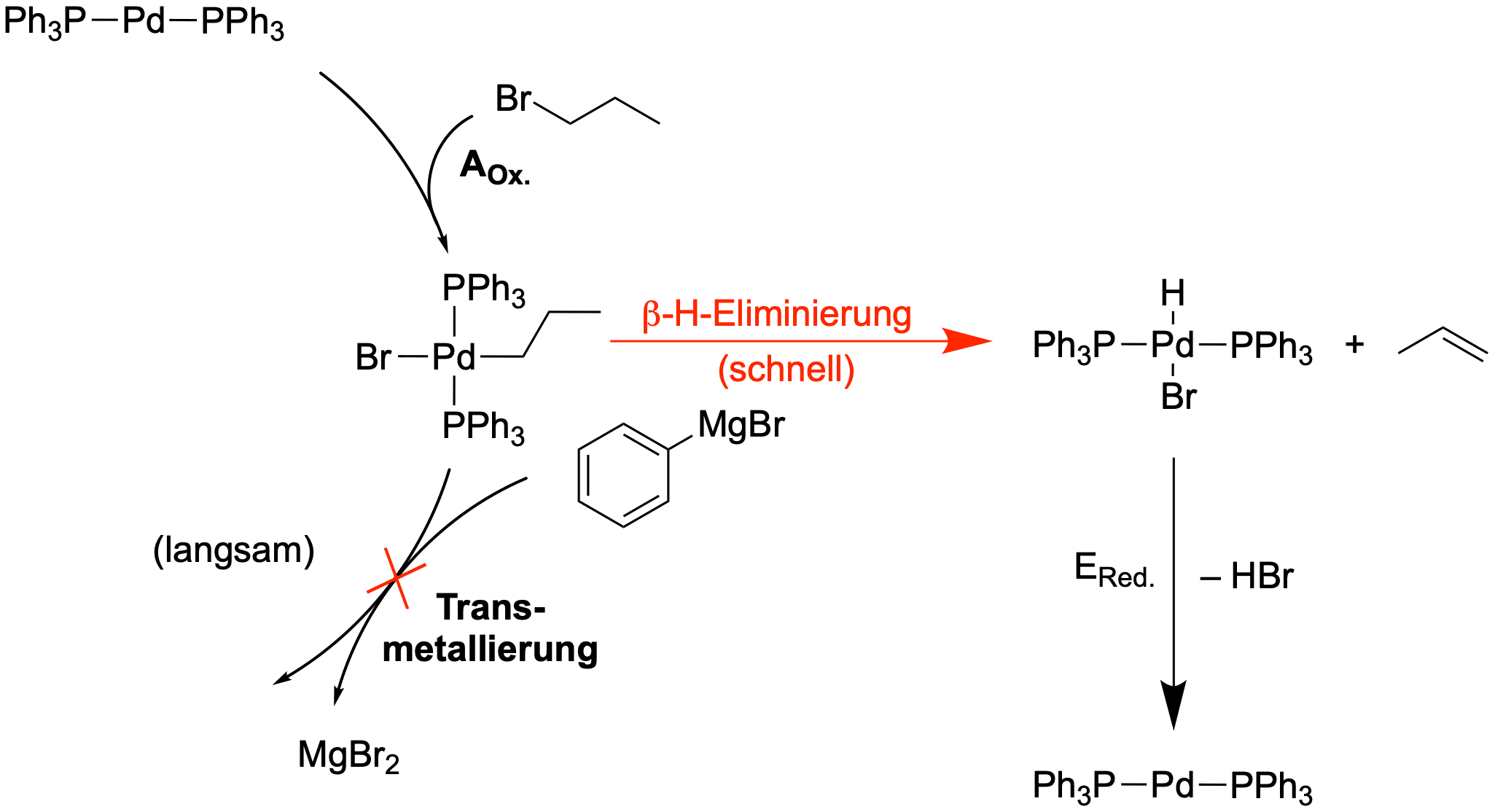
Um dies zu verhindern, darf das Elektrophil über keinen sp3-Kohlenstoff in β-Position besitzen, welches mind. ein Wasserstoff bindet (R= Ethyl, Isobutyl, etc.). Möglich hingegen sind bspw. die Reste R= Allyl, Vinyl, Aryl, Alkinyl...
Weitere Kreuzkupplungen[1]
Es gibt auch viele Kreuzkupplungen, welche von diesem Standardschema leicht abweichen, um andere Arten von Reaktionen zu realisieren. Wir schauen uns jetzt noch die Sonogashira-, Buchwald-Hartwig- und die Heck-Kreuzkupplung an.Die Sonogashira-Kupplung
Bei der Sonogashira-Kupplung wird das Nucleophil in situ dargestellt, indem ein terminales Alkin unter Kupfer(I)-Katalyse deprotoniert wird um ein Cuprat im Reaktionsmedium darzustellen, welches die Transmetallierung durchführt. So gibt es zwei Katalysezyklen, welche sich am Punkt der Transmetallierung berühren. Der Kupfer-Zyklus wird auch Glaser-Zyklus genannt, als Anlehung an die Glaser-Homokupplung von Alkinen, welche ebenfalls unter Kupfer-Katalyse verläuft.
Warum kann das Alkin so leicht deprotoniert werden?
Außerdem wird durch den π-Komplex, welches das Alkin hier mit dem Cu(I)-Katalysator bildet, die Acidität des Alkins noch weiter erhöht, da diesem Elektronendichte entzogen wird. Das erleichtert die Deprotonierung zusätzlich.
Die Buchwald-Hartwig-Kupplung
Bei dieser Kreuzkupplung wird ein Amin als Nucleophil eingesetzt, weshalb es sich hier um eine Aminierung handelt. Hier wird sogar von einem einach koordinierten Komplex ausgegangen, damit die Reaktion wie folgt durchlaufen werden kann:
Interessant ist, dass der Stickstoff durch die Deprotonierung von einem neutralen Liganden in einen anionischen Liganden überführt wird.
Es kann auch zu Nebenreaktionen durch beta-H-Eliminierungen kommen, falls die Reste am Amine Alkylreste sind:
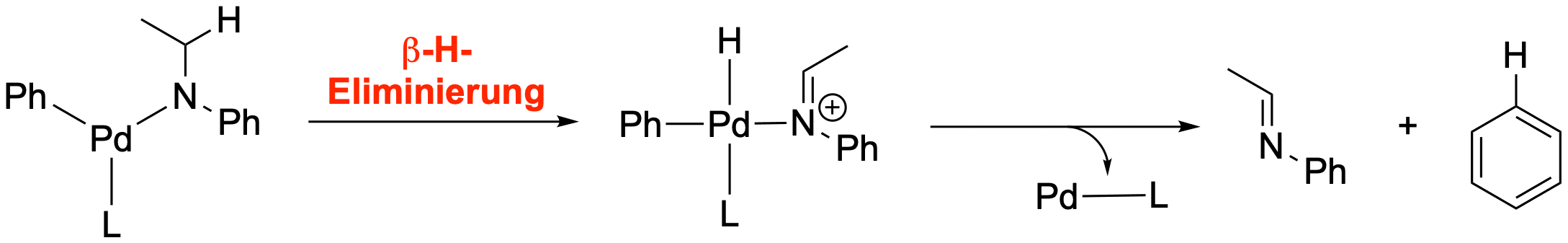
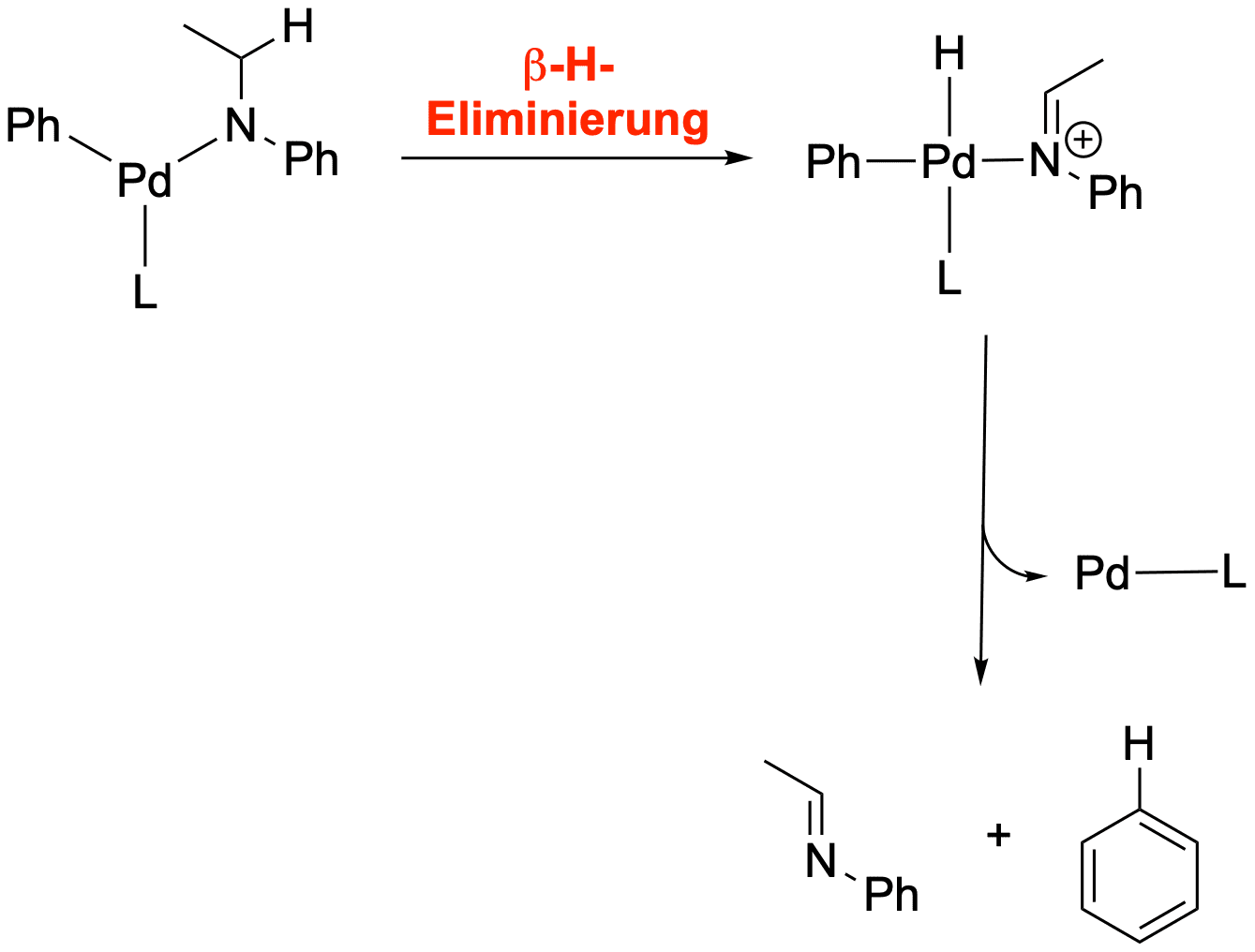
Die Heck-Kupplung[3,4]
Zum Abschluss wollen wir eine mechanistisch etwas unterschiedliche Kreuzkupplung anschauen. Ein Nucleophil gibt es bei dieser Reaktion in diesem Sinne nicht. Als Gegenstück zum Kupplungspartner R-X wird nämlich ein Alken mit mindestens einem Wasserstoff benötigt.
Anstelle einer Transmetallierung findet hier eine Carbometallierung statt.
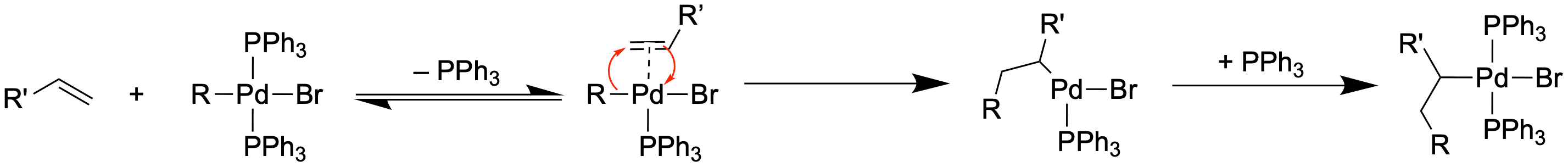
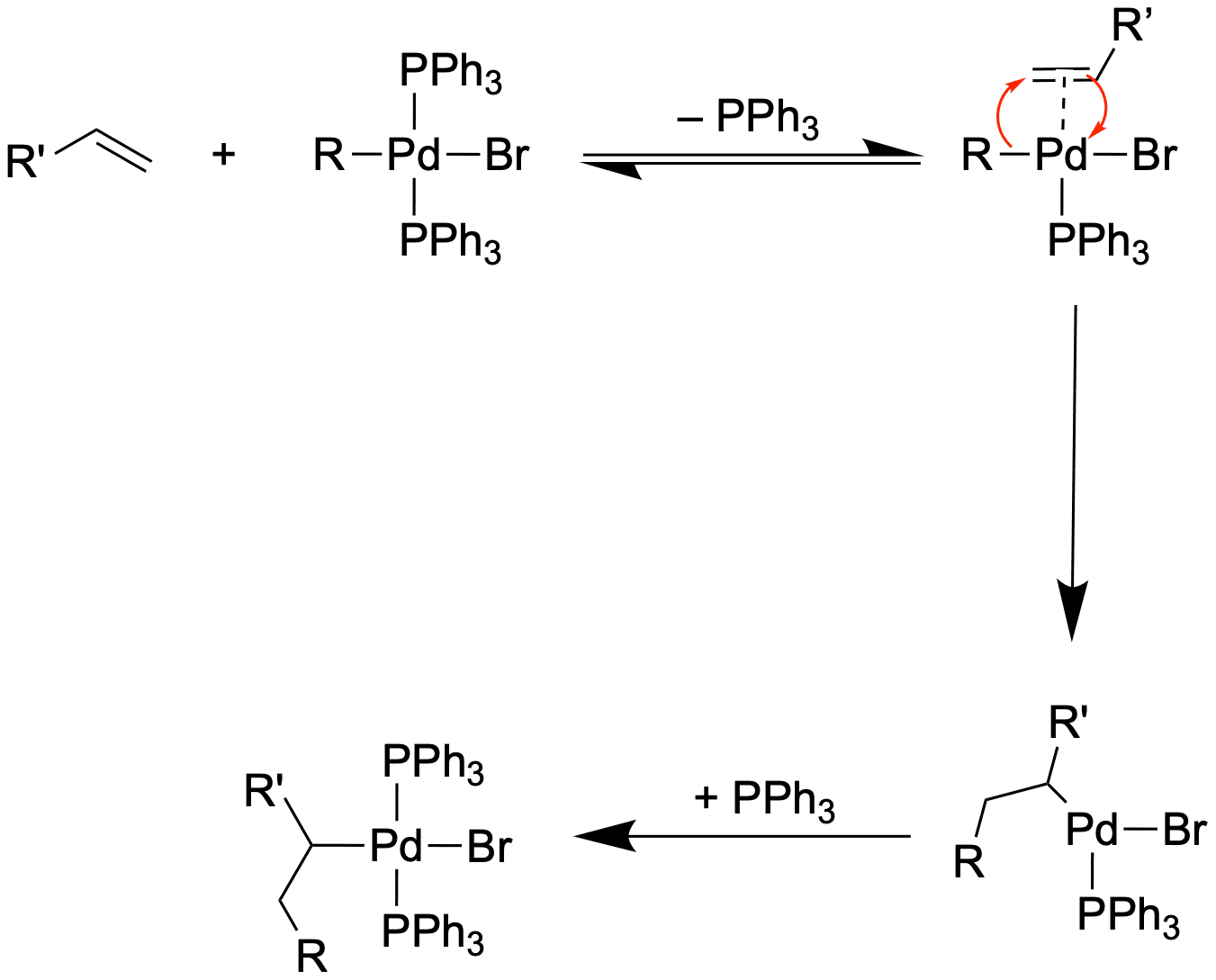
Die Carbometallierung kann mit einer Sequenz von drei Reaktionen beschrieben werden:
- Ligandenaustausch zur Bildung des π-Komplex
- Insertion in die R-M-Bindung
C-M-Bindungen sind sehr reaktiv gegenüber C-C-Doppelbindungen. Diese Reaktion kennst du vielleicht bereits durch die Ziegler-Natta-Katalysatoren - Sättigung der Koordinationssphäre
Auf die Carbometallierung folgt eine β-Hydrid-Eliminierung, welche die C-C-Doppelbindung wiederherstellt. Auf diese Weise haben wir ein Michael-System in β-Position substituiert - sehr nützlich, oder?
Auf einem Blick...
Katalysator
• Meistens Pd(0)
• Wechselt zwischen Pd(0) & Pd(II)
• Pd(II) koordiniert als d8-System quadratisch Planar
• Dissoziation von zwei Liganden zur Bildung der reaktiven Katalysatorspezies
• Pd(II)-Verbindungen als Precursor
Kreuzkupplungen
• AOx, Transmetallierung, Isomerisierung, ERed
• Nucleophil variabel (Kumada, Negishi, Stile, Suzuki...)
• Toleranz gegenüber FG steigt mit höheren kovalenten Charkakter der R-M-Bindung
• β-H-Eliminierungen und Carbometallierungen als weitere prominente Reaktionen
Referenzen
1. J.Clayden, N.Greeves, S.Warren, P.Wothers in Organic Chemistry, Vol. 8, Oxford University Press, Oxford, 2008.
2. A. Suzuki, J. Organomet. Chem. 1999, 576, 147-168.
3. R. Heck, J. Nolley, J. Org. Chem. 1972, 37, 2320-2322.
4. G.Crisp, Chem. Soc. Rev. 1998, 27, 427-436.
Wenn nicht anders angegeben, sind alle Abbildungen selbst angefertigt.